
Nasal Turbinate Mesenchymal Stromal Cells Preserve Characteristics of Their Neural Crest Origin and Exert Distinct Paracrine Activity

 ,
,  , and
, and
Abstract
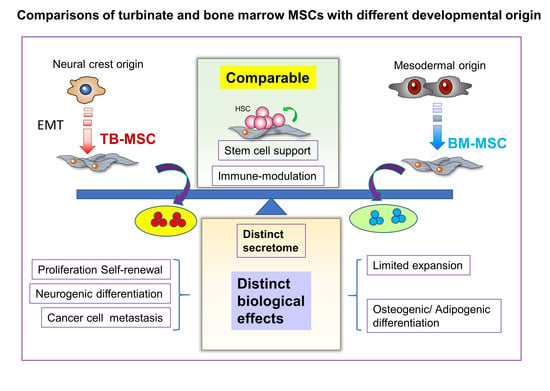
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Isolation of Cells and Culture
2.2. Phenotyping of MSCs
2.3. Colony-Formation Assay
2.4. Multilineage Differentiation Assays
2.5. RNA Extraction and qRT-PCR
2.6. Growth Factors for Cell Stimulation
2.7. Co-Culture
2.8. Neural Differentiation
2.9. Immunocytochemistry and Western Blotting
2.10. Tumor Growth and Metastasis
2.11. Immune Suppression Assay
2.12. Mass Spectrometric (MS) Analysis of Secreted Proteins
2.13. Bioinformatic Analysis of Mass Spectrometric Data
2.14. Statistical Analysis
3. Results
3.1. Turbinate and Bone Marrow MSCs Exhibit Distinct Cellular Characteristics
3.2. Turbinate MSCs Preserve the Neural Crest-Like Properties in Cell Autonomous Effects
3.3. Proteomic Distinctions of TB and BM-MSCs in Their Secreted Proteins
3.4. Comparisons of Paracrine Effects of TB and BM-MSCs on Normal and Cancer Stem Cells
3.5. Comparisons for Immune-Modulating Effects of TB and BM-MSCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Keating, A. Mesenchymal stromal cells. Curr. Opin. Hematol. 2006, 13, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prockop, D.J. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 1997, 276, 71–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões, I.N.; Boura, J.S.; dos Santos, F.; Andrade, P.Z.; Cardoso, C.M.; Gimble, J.M.; da Silva, C.L.; Cabral, J.M. Human mesenchymal stem cells from the umbilical cord matrix: Successful isolation and ex vivo expansion using serum-/xeno-free culture media. Biotechnol. J. 2013, 8, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Galderisi, U.; Marino, I.R. From the laboratory bench to the patient’s bedside: An update on clinical trials with mesenchymal stem cells. J. Cell. Physiol. 2007, 211, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Dezawa, M.; Hoshino, M.; Ide, C. Treatment of neurodegenerative diseases using adult bone marrow stromal cell-derived neurons. Expert Opin. Biol. Ther. 2005, 5, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Gang, E.J.; Jeong, J.A.; Hong, S.H.; Hwang, S.H.; Kim, S.W.; Yang, I.H.; Ahn, C.; Han, H.; Kim, H. Skeletal myogenic differentiation of mesenchymal stem cells isolated from human umbilical cord blood. Stem Cells 2004, 22, 617–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwitz, E.M.; Gordon, P.L.; Koo, W.K.; Marx, J.C.; Neel, M.D.; McNall, R.Y.; Muul, L.; Hofmann, T. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implications for cell therapy of bone. Proc. Natl. Acad. Sci. USA 2002, 99, 8932–8937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, C.; Gordeladze, J.; Noel, D. Tissue engineering through autologous mesenchymal stem cells. Curr. Opin. Biotechnol. 2004, 15, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Magne, D.; Vinatier, C.; Julien, M.; Weiss, P.; Guicheux, J. Mesenchymal stem cell therapy to rebuild cartilage. Trends Mol. Med. 2005, 11, 519–526. [Google Scholar] [CrossRef]
- Krampera, M.; Glennie, S.; Dyson, J.; Scott, D.; Laylor, R.; Simpson, E.; Dazzi, F. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood 2002, 101, 3722–3729. [Google Scholar] [CrossRef]
- Le Blanc, K.; Tammik, L.; Sundberg, B.; Haynesworth, S.E.; Ringden, O. Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand. J. Immunol. 2003, 57, 11–20. [Google Scholar] [CrossRef]
- Kim, D.W.; Chung, Y.J.; Kim, T.G.; Kim, Y.L.; Oh, I.H. Cotransplantation of third-party mesenchymal stromal cells can alleviate single-donor predominance and increase engraftment from double cord transplantation. Blood 2004, 103, 1941–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noort, W.A.; Kruisselbrink, A.B.; in’t Anker, P.S.; Kruger, M.; van Bezooijen, R.L.; de Paus, R.A.; Heemskerk, M.H.; Lowik, C.W.; Falkenburg, J.H.; Willemze, R.; et al. Mesenchymal stem cells promote engraftment of human umbilical cord blood-derived CD34(+) cells in NOD/SCID mice. Exp. Hematol. 2002, 30, 870–878. [Google Scholar] [CrossRef]
- Le Blanc, K.; Rasmusson, I.; Sundberg, B.; Gotherstrom, C.; Hassan, M.; Uzunel, M.; Ringden, O. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet 2004, 363, 1439–1441. [Google Scholar] [CrossRef]
- Ringden, O.; Uzunel, M.; Rasmusson, I.; Remberger, M.; Sundberg, B.; Lonnies, H.; Marschall, H.U.; Dlugosz, A.; Szakos, A.; Hassan, Z.; et al. Mesenchymal stem cells for treatment of therapy-resistant graft-versus-host disease. Transplantation 2006, 81, 1390–1397. [Google Scholar] [CrossRef]
- Colter, D.C.; Sekiya, I.; Prockop, D.J. Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc. Natl. Acad. Sci. USA 2001, 98, 7841–7845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucia, M.; Halasa, M.; Wsoczynski, M.; Baskiewicz-Masiuk, M.; Zuba-Surma, E.; Machalinski, B.; Ratajczak, M.Z. A novel population of Oct-4+ SSEA-1+ CXCR4+ CD34+ CD133+ Lin- CD45- very smal embryonic-like (VSEL) stem cells identified in human cord blood. Blood 2006, 108, 912a. [Google Scholar] [CrossRef]
- Smith, J.R.; Pochampally, R.; Perry, A.; Hsu, S.C.; Prockop, D.J. Isolation of a highly clonogenic and multipotential subfraction of adult stem cells from bone marrow stroma. Stem Cells 2004, 22, 823–831. [Google Scholar] [CrossRef]
- Lee, G.Y.; Jeong, S.Y.; Lee, H.R.; Oh, I.H. Age-related differences in the bone marrow stem cell niche generate specialized microenvironments for the distinct regulation of normal hematopoietic and leukemia stem cells. Sci. Rep. 2019, 9, 1007. [Google Scholar] [CrossRef] [PubMed]
- Burja, B.; Barlič, A.; Erman, A.; Mrak-Poljšak, K.; Tomšič, M.; Sodin-Semrl, S.; Lakota, K. Human mesenchymal stromal cells from different tissues exhibit unique responses to different inflammatory stimuli. Curr. Res. Transl. Med. 2020, 68, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Kasper, C.; Böhm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal. 2011, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, S.; Eichler, H.; Stoeve, J.; Klüter, H.; Bieback, K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006, 24, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, H.S.; Choi, H.K.; Kim, J.A.; Chu, I.S.; Leem, S.H.; Oh, I.H. Heterogeneous Niche Activity of Ex-Vivo Expanded MSCs as Factor for Variable Outcomes in Hematopoietic Recovery. PLoS ONE 2016, 11, e0168036. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.; Widera, D.; Qunneis, F.; Müller, J.; Zander, C.; Greiner, J.; Strauss, C.; Lüningschrör, P.; Heimann, P.; Schwarze, H.; et al. Isolation of novel multipotent neural crest-derived stem cells from adult human inferior turbinate. Stem Cells Dev. 2012, 21, 742–756. [Google Scholar] [CrossRef]
- Tomé, M.; Lindsay, S.L.; Riddell, J.S.; Barnett, S.C. Identification of nonepithelial multipotent cells in the embryonic olfactory mucosa. Stem Cells 2009, 27, 2196–2208. [Google Scholar] [CrossRef] [PubMed]
- Murrell, W.; Féron, F.; Wetzig, A.; Cameron, N.; Splatt, K.; Bellette, B.; Bianco, J.; Perry, C.; Lee, G.; Mackay-Sim, A. Multipotent stem cells from adult olfactory mucosa. Dev. Dyn. 2005, 233, 496–515. [Google Scholar] [CrossRef]
- Trentin, A.; Glavieux-Pardanaud, C.; Le Douarin, N.M.; Dupin, E. Self-renewal capacity is a widespread property of various types of neural crest precursor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4495–4500. [Google Scholar] [CrossRef] [Green Version]
- Stemple, D.L.; Anderson, D.J. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell 1992, 71, 973–985. [Google Scholar] [CrossRef]
- Nagoshi, N.; Shibata, S.; Kubota, Y.; Nakamura, M.; Nagai, Y.; Satoh, E.; Morikawa, S.; Okada, Y.; Mabuchi, Y.; Katoh, H.; et al. Ontogeny and multipotency of neural crest-derived stem cells in mouse bone marrow, dorsal root ganglia, and whisker pad. Cell Stem Cell 2008, 2, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.; Moon, N.; Ahn, J.Y.; Oh, E.J.; Kim, M.; Cho, C.S.; Shin, J.C.; Oh, I.H. Mesenchymal stromal cells expanded in human allogenic cord blood serum display higher self-renewal and enhanced osteogenic potential. Stem Cells Dev. 2009, 18, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.C.; Chuu, C.P.; Chen, C.Y.; Shiah, S.G.; Kung, H.J.; King, K.L.; Su, L.C.; Chang, S.C.; Chang, C.H. Elevation of soluble guanylate cyclase suppresses proliferation and survival of human breast cancer cells. PLoS ONE 2015, 10, e0125518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.; Lee, J.; Kwon, Y.; Park, K.S.; Jeong, J.H.; Choi, S.J.; Bang, S.I.; Chang, J.W.; Lee, C. Comparative Proteomic Analysis of the Mesenchymal Stem Cells Secretome from Adipose, Bone Marrow, Placenta and Wharton’s Jelly. Int. J. Mol. Sci. 2021, 22, 845. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Kwon, Y.; Lee, S.; Na, S.; Hong, E.Y.; Ju, S.; Jung, H.G.; Kaushal, P.; Shin, S.; Back, J.H.; et al. Common Repository of FBS Proteins (cRFP) To Be Added to a Search Database for Mass Spectrometric Analysis of Cell Secretome. J. Proteome Res. 2019, 18, 3800–3806. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Bendtsen, J.D.; Jensen, L.J.; Blom, N.; Von Heijne, G.; Brunak, S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. 2004, 17, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Möller, S.; Croning, M.D.; Apweiler, R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 2001, 17, 646–653. [Google Scholar] [CrossRef] [Green Version]
- da Silva Meirelles, L.; de Deus Wagatsuma, V.M.; Malta, T.M.; Bonini Palma, P.V.; Araújo, A.G.; Panepucci, R.A.; Silva, W.A.; Kashima, S.; Covas, D.T. The gene expression profile of non-cultured, highly purified human adipose tissue pericytes: Transcriptomic evidence that pericytes are stem cells in human adipose tissue. Exp. Cell Res. 2016, 349, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.C.D.; Rustenhoven, J.; Scotter, E.L.; Schweder, P.; Faull, R.L.M.; Park, T.I.H.; Dragunow, M. Markers for human brain pericytes and smooth muscle cells. J. Chem. Neuroanat. 2018, 92, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Boudjadi, S.; Chatterjee, B.; Sun, W.; Vemu, P.; Barr, F.G. The expression and function of PAX3 in development and disease. Gene 2018, 666, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Isern, J.; García-García, A.; Martín, A.M.; Arranz, L.; Martín-Pérez, D.; Torroja, C.; Sánchez-Cabo, F.; Méndez-Ferrer, S. The neural crest is a source of mesenchymal stem cells with specialized hematopoietic stem cell niche function. eLife 2014, 3, e03696. [Google Scholar] [CrossRef] [PubMed]
- Shafit-Zagardo, B.; Kalcheva, N. Making sense of the multiple MAP-2 transcripts and their role in the neuron. Mol. Neurobiol. 1998, 16, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.L.; Leube, R.E.; Cousin, M.A. Synaptophysin is required for synaptobrevin retrieval during synaptic vesicle endocytosis. J. Neurosci. 2011, 31, 14032–14036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarsa, L.; Goda, Y. Synaptophysin regulates activity-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA 2002, 99, 1012–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucchi, E.; Bonetto, V.; Sorarù, G.; Martinelli, I.; Parchi, P.; Liguori, R.; Mandrioli, J. Neurofilaments in motor neuron disorders: Towards promising diagnostic and prognostic biomarkers. Mol. Neurodegener. 2020, 15, 58. [Google Scholar] [CrossRef] [PubMed]
- Mullen, R.J.; Buck, C.R.; Smith, A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Development 1992, 116, 201–211. [Google Scholar] [PubMed]
- Murray, L.; Chen, B.; Galy, A.; Chen, S.; Tushinski, R.; Uchida, N.; Negrin, R.; Tricot, G.; Jagannath, S.; Vesole, D.; et al. Enrichment of human hematopoietic stem cell activity in the CD34+Thy-1+Lin- subpopulation from mobilized peripheral blood. Blood 1995, 85, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Péault, B.; Weissman, I.L.; Buckle, A.M.; Tsukamoto, A.; Baum, C. Thy-1-expressing CD34+ human cells express multiple hematopoietic potentialities in vitro and in SCID-hu mice. Nouv. Rev. Fr. Hematol. 1993, 35, 91–93. [Google Scholar]
- Jeong, S.Y.; Kim, J.A.; Oh, I.H. The Adaptive Remodeling of Stem Cell Niche in Stimulated Bone Marrow Counteracts the Leukemic Niche. Stem Cells 2018, 36, 1617–1629. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.A.; Shim, J.S.; Lee, G.Y.; Yim, H.W.; Kim, T.M.; Kim, M.; Leem, S.H.; Lee, J.W.; Min, C.K.; Oh, I.H. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015, 75, 2222–2231. [Google Scholar] [CrossRef] [Green Version]
- Oh, I.H.; Kwon, K.R. Concise review: Multiple niches for hematopoietic stem cell regulations. Stem Cells 2010, 28, 1243–1249. [Google Scholar]
- Dittmer, J.; Rody, A. Cancer stem cells in breast cancer. Histol. Histopathol. 2013, 28, 827–838. [Google Scholar] [PubMed]
- Velasco-Velázquez, M.A.; Popov, V.M.; Lisanti, M.P.; Pestell, R.G. The role of breast cancer stem cells in metastasis and therapeutic implications. Am. J. Pathol. 2011, 179, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Czapla, J.; Matuszczak, S.; Kulik, K.; Wiśniewska, E.; Pilny, E.; Jarosz-Biej, M.; Smolarczyk, R.; Sirek, T.; Zembala, M.O.; Zembala, M.; et al. The effect of culture media on large-scale expansion and characteristic of adipose tissue-derived mesenchymal stromal cells. Stem Cell Res. Ther. 2019, 10, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, G. The developmental basis of mesenchymal stem/stromal cells (MSCs). BMC Dev. Biol. 2015, 15, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretlow, J.D.; Jin, Y.Q.; Liu, W.; Zhang, W.J.; Hong, T.H.; Zhou, G.; Baggett, L.S.; Mikos, A.G.; Cao, Y. Donor age and cell passage affects differentiation potential of murine bone marrow-derived stem cells. BMC Cell Biol. 2008, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Wagner, W.; Bork, S.; Horn, P.; Krunic, D.; Walenda, T.; Diehlmann, A.; Benes, V.; Blake, J.; Huber, F.X.; Eckstein, V.; et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS ONE 2009, 4, e5846. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Wang, Y.M.; Zhang, H.; Li, J.; Wang, W.; Wei, Y.J.; Hu, S.S. Aging adversely impacts biological properties of human bone marrow-derived mesenchymal stem cells: Implications for tissue engineering heart valve construction. Artif. Organs 2010, 34, 215–222. [Google Scholar] [CrossRef]
- Viswanathan, S.; Keating, A.; Deans, R.; Hematti, P.; Prockop, D.; Stroncek, D.F.; Stacey, G.; Weiss, D.J.; Mason, C.; Rao, M.S. Soliciting strategies for developing cell-based reference materials to advance mesenchymal stromal cell research and clinical translation. Stem Cells Dev. 2014, 23, 1157–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials With Mesenchymal Stem Cells: An Update. Cell Transplant. 2016, 25, 829–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaim, M.; Karaman, S.; Cetin, G.; Isik, S. Donor age and long-term culture affect differentiation and proliferation of human bone marrow mesenchymal stem cells. Ann. Hematol. 2012, 91, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Horn, P.; Castoldi, M.; Diehlmann, A.; Bork, S.; Saffrich, R.; Benes, V.; Blake, J.; Pfister, S.; Eckstein, V.; et al. Replicative senescence of mesenchymal stem cells: A continuous and organized process. PLoS ONE 2008, 3, e2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, D.J. Lineages and transcription factors in the specification of vertebrate primary sensory neurons. Curr. Opin. Neurobiol. 1999, 9, 517–524. [Google Scholar] [CrossRef]
- Korzh, V.; Strähle, U. Proneural, prosensory, antiglial: The many faces of neurogenins. Trends Neurosci. 2002, 25, 603–605. [Google Scholar] [CrossRef]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Lester, B.R.; McCarthy, J.B. Tumor cell adhesion to the extracellular matrix and signal transduction mechanisms implicated in tumor cell motility, invasion and metastasis. Cancer Metastasis Rev. 1992, 11, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Schinelli, S. Extracellular Matrix Alterations in Metastatic Processes. Int. J. Mol. Sci. 2019, 20, 4947. [Google Scholar] [CrossRef] [Green Version]
- Stetler-Stevenson, W.G.; Aznavoorian, S.; Liotta, L.A. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu. Rev. Cell Biol. 1993, 9, 541–573. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-J.; Shin, S.; Jeong, S.-Y.; Lim, S.-U.; Lee, D.-W.; Kwon, Y.-K.; Kang, J.; Kim, S.-W.; Jung, C.-K.; Lee, C.; et al. Nasal Turbinate Mesenchymal Stromal Cells Preserve Characteristics of Their Neural Crest Origin and Exert Distinct Paracrine Activity. J. Clin. Med. 2021, 10, 1792. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10081792
Kim H-J, Shin S, Jeong S-Y, Lim S-U, Lee D-W, Kwon Y-K, Kang J, Kim S-W, Jung C-K, Lee C, et al. Nasal Turbinate Mesenchymal Stromal Cells Preserve Characteristics of Their Neural Crest Origin and Exert Distinct Paracrine Activity. Journal of Clinical Medicine. 2021; 10(8):1792. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10081792
Chicago/Turabian StyleKim, Hyun-Jee, Sungho Shin, Seon-Yeong Jeong, Sun-Ung Lim, Dae-Won Lee, Yunhee-Kim Kwon, Jiyeon Kang, Sung-Won Kim, Chan-Kwon Jung, Cheolju Lee, and et al. 2021. "Nasal Turbinate Mesenchymal Stromal Cells Preserve Characteristics of Their Neural Crest Origin and Exert Distinct Paracrine Activity" Journal of Clinical Medicine 10, no. 8: 1792. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10081792