
Advanced Microscopy for Liver and Gut Ultrastructural Pathology in Patients with MVID and PFIC Caused by MYO5B Mutations

 , , , and
, , , and
Abstract
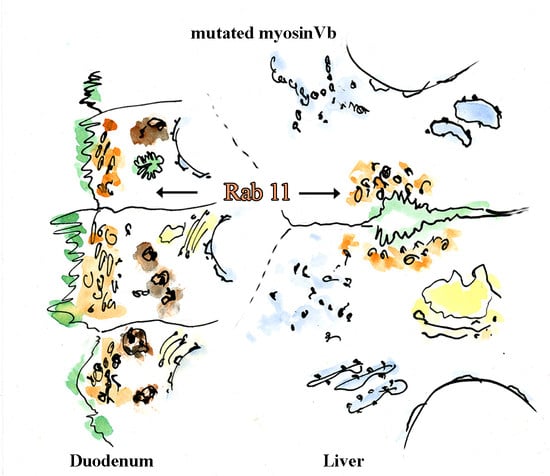
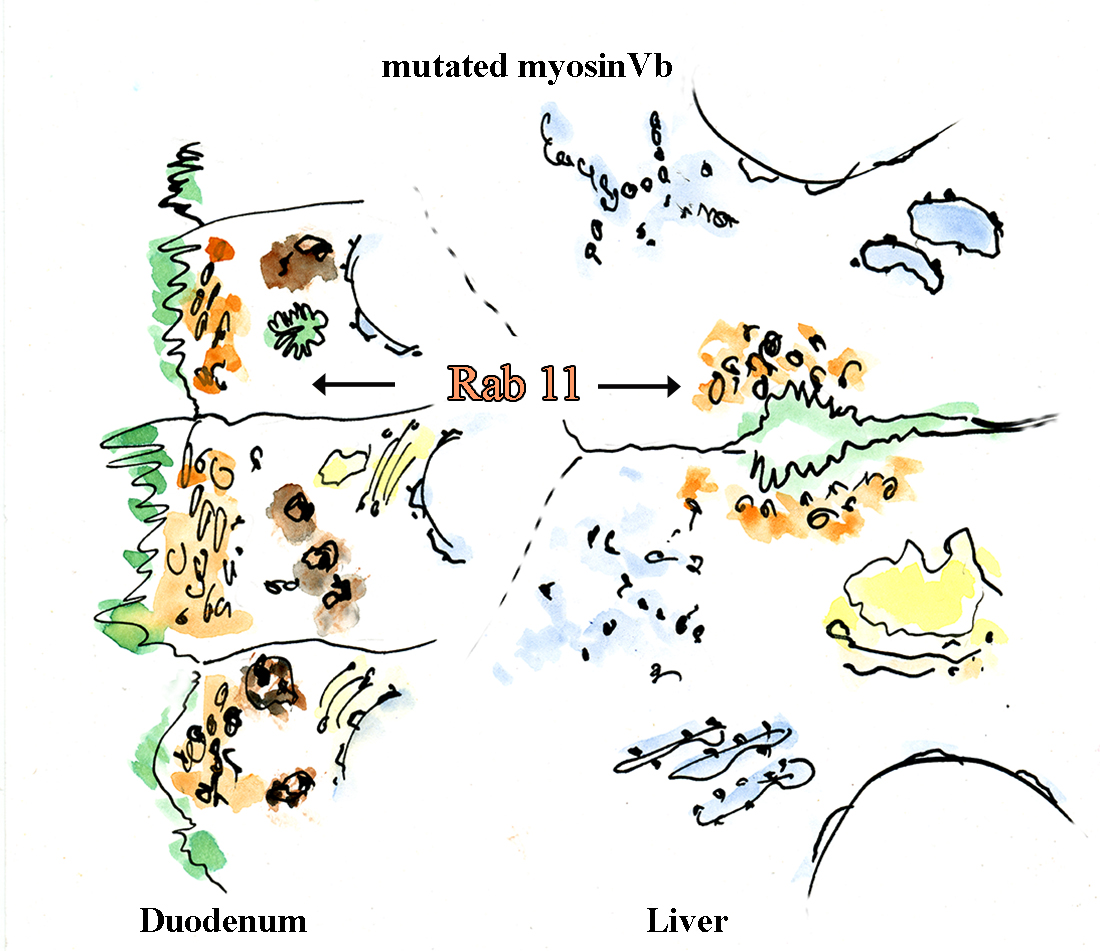
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Methodological Rationale
1.2. Disease Investigated
1.3. Aim of This Study
2. Materials and Methods
2.1. Biopsy Sampling
2.2. Enteroid Culture
2.2.1. Reagents
2.2.2. Generation and Culture of Patient Enteroids
2.3. Fluorescence Microscopy (IF)
2.4. Ultrastructural Analyses (EM Approaches)
2.4.1. Reagents
2.4.2. Specimen Preparation
- (a)
- Chemical fixation
- (b)
- Standard processing for resin embedding
- (c)
- (d)
- Ultramicrotomy, staining and PAS cytochemistry of resin blocks
- (e)
- Tokuyasu immuno-EM [20]
2.4.3. Electron Microscopy and Electron Tomography for 2D and 3D Analyses, Respectively
2.5. Cell Culture and Immunoblot Analysis to Validate Anti-CFTR Monoclonal Antibody #596
2.6. Study Approval
3. Results
3.1. Clinical Data—Patient Description
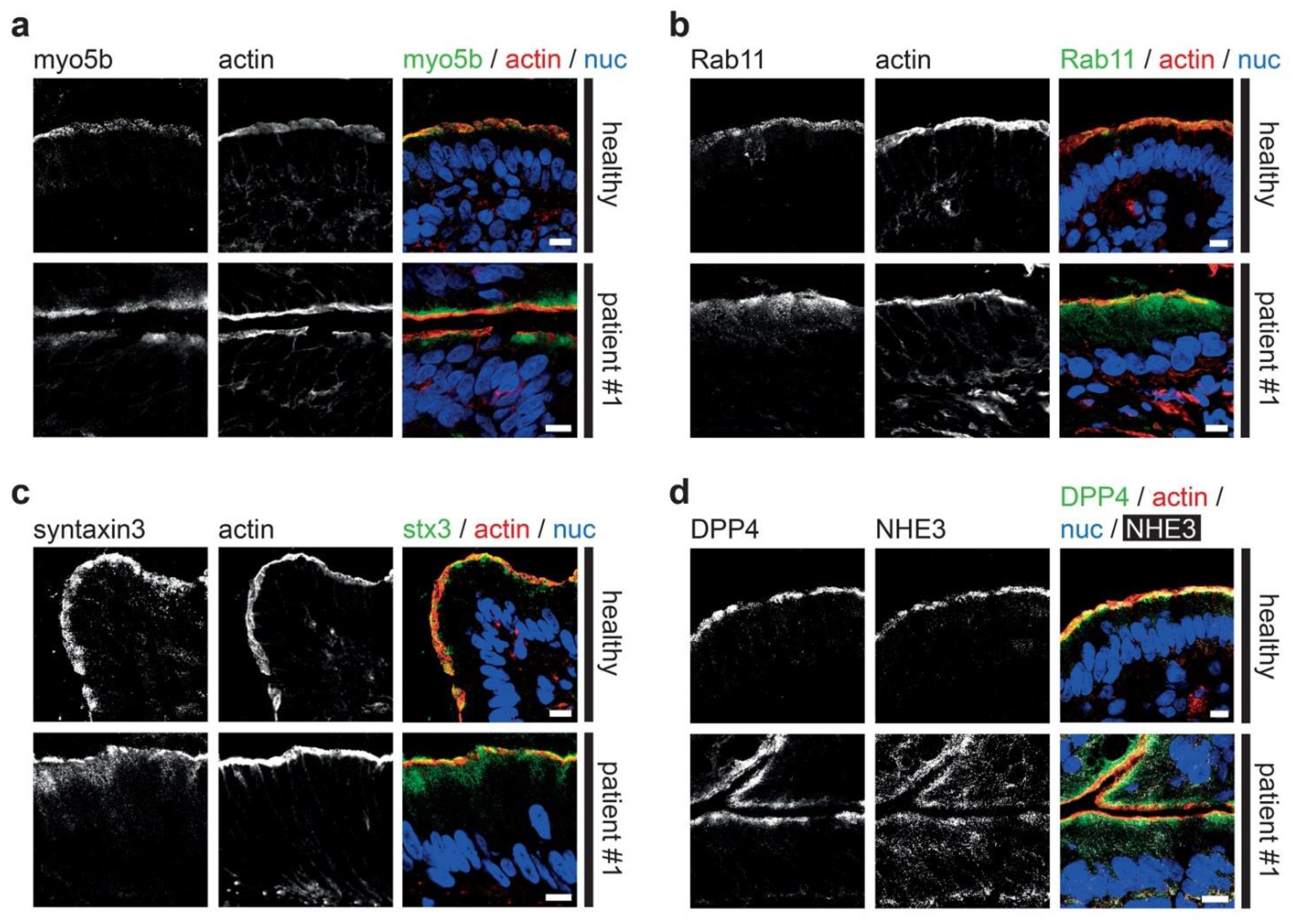
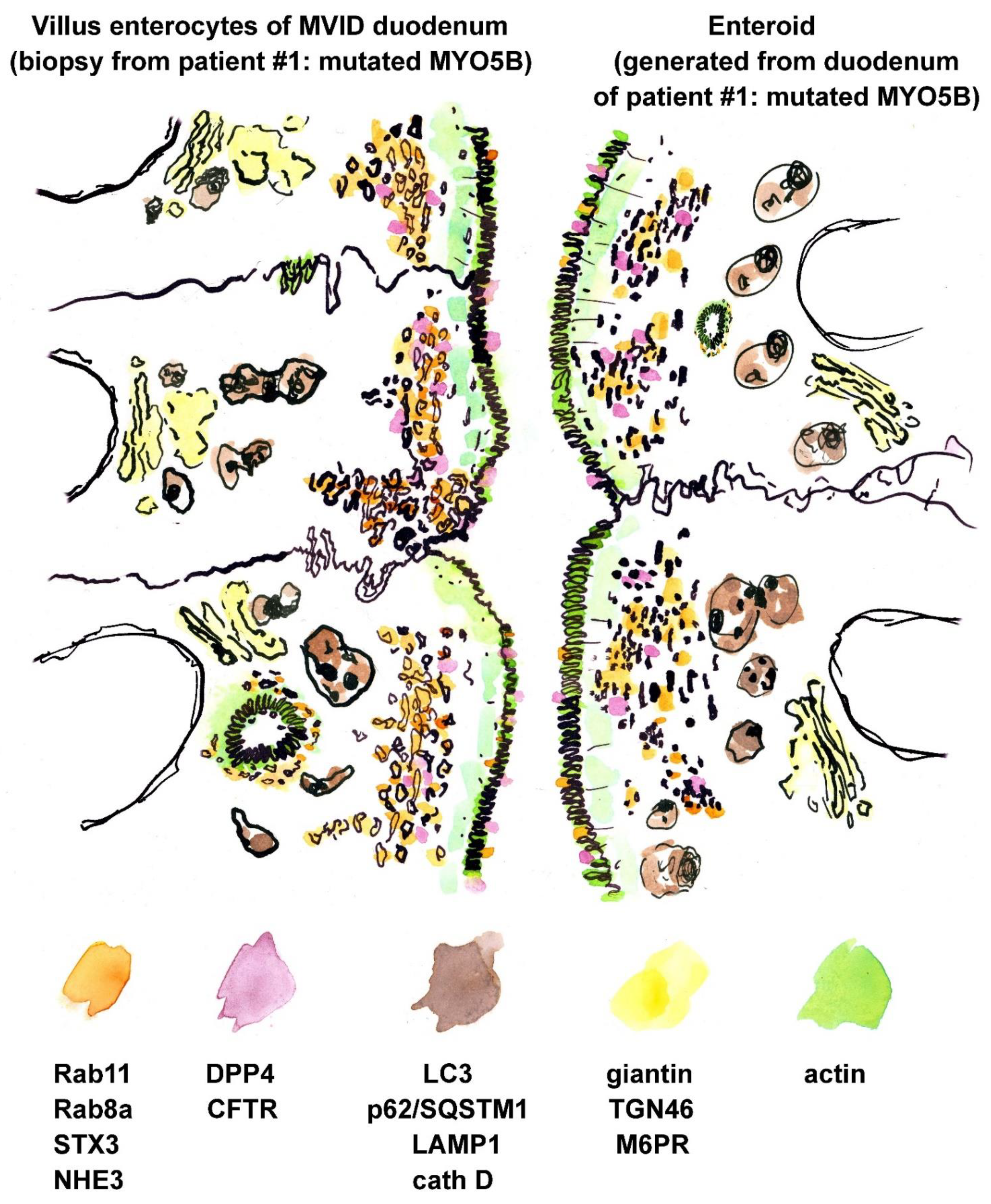
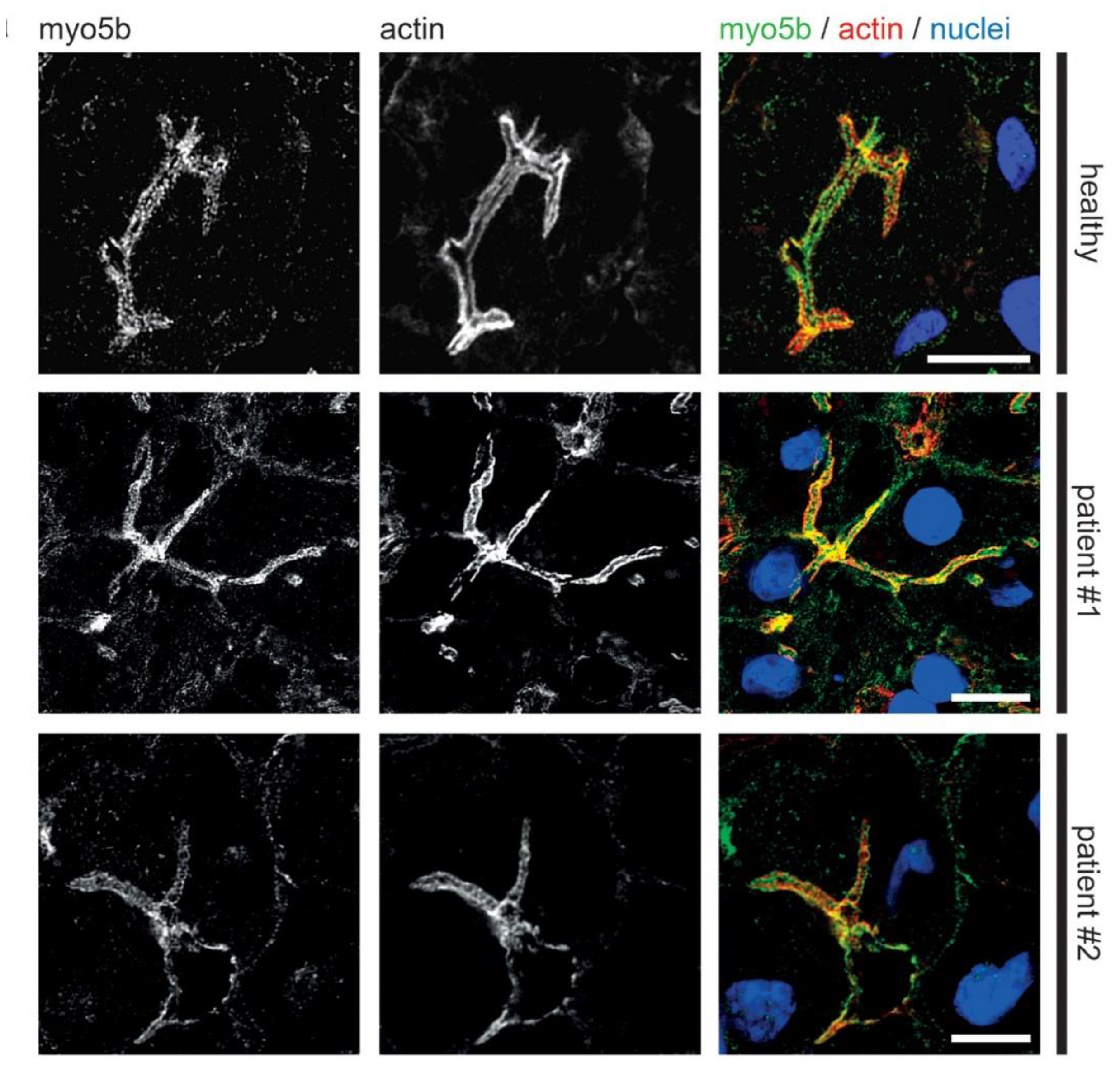
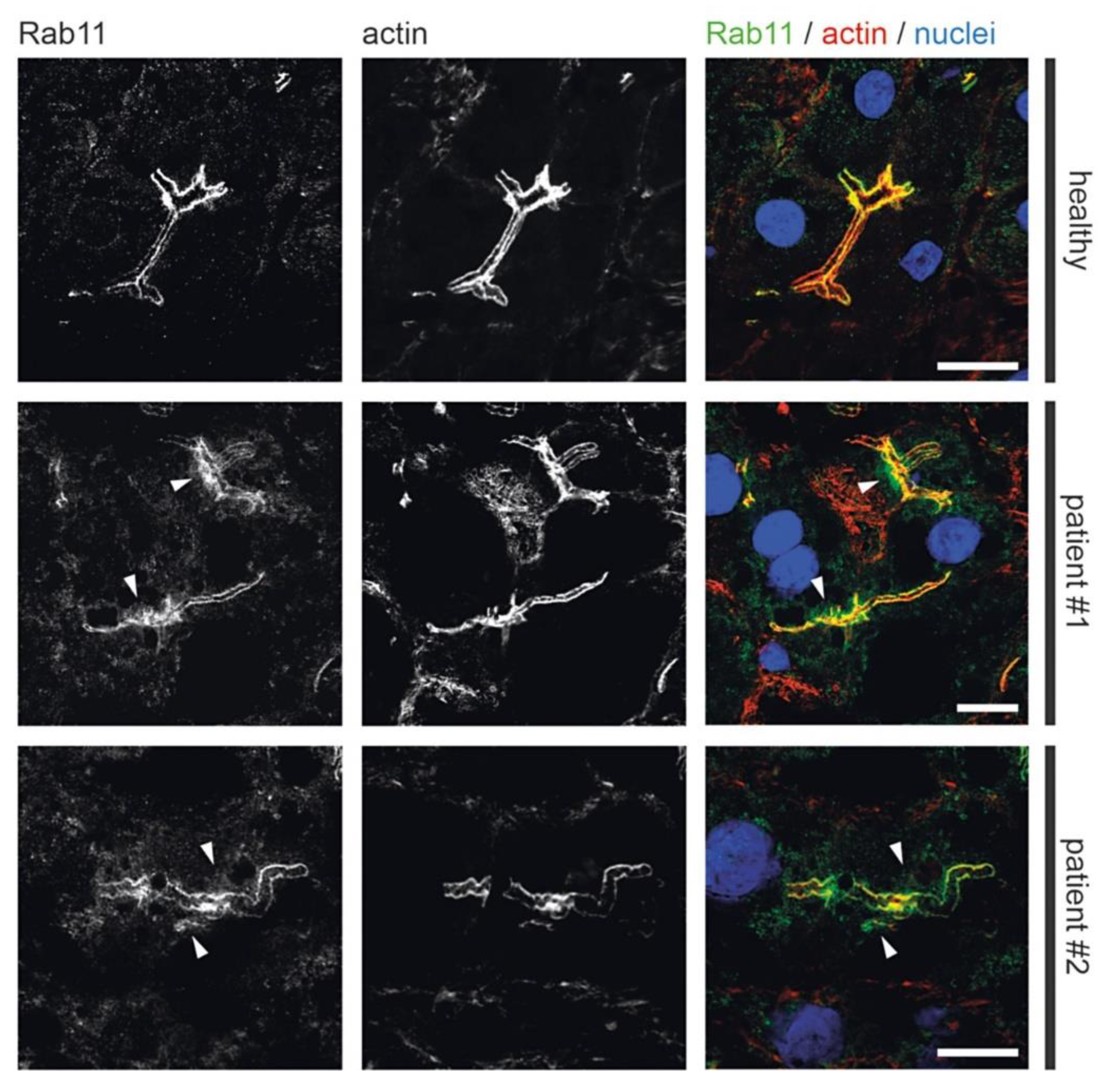
3.2. Fluorescence Microcopy of Duodenal Biopsies
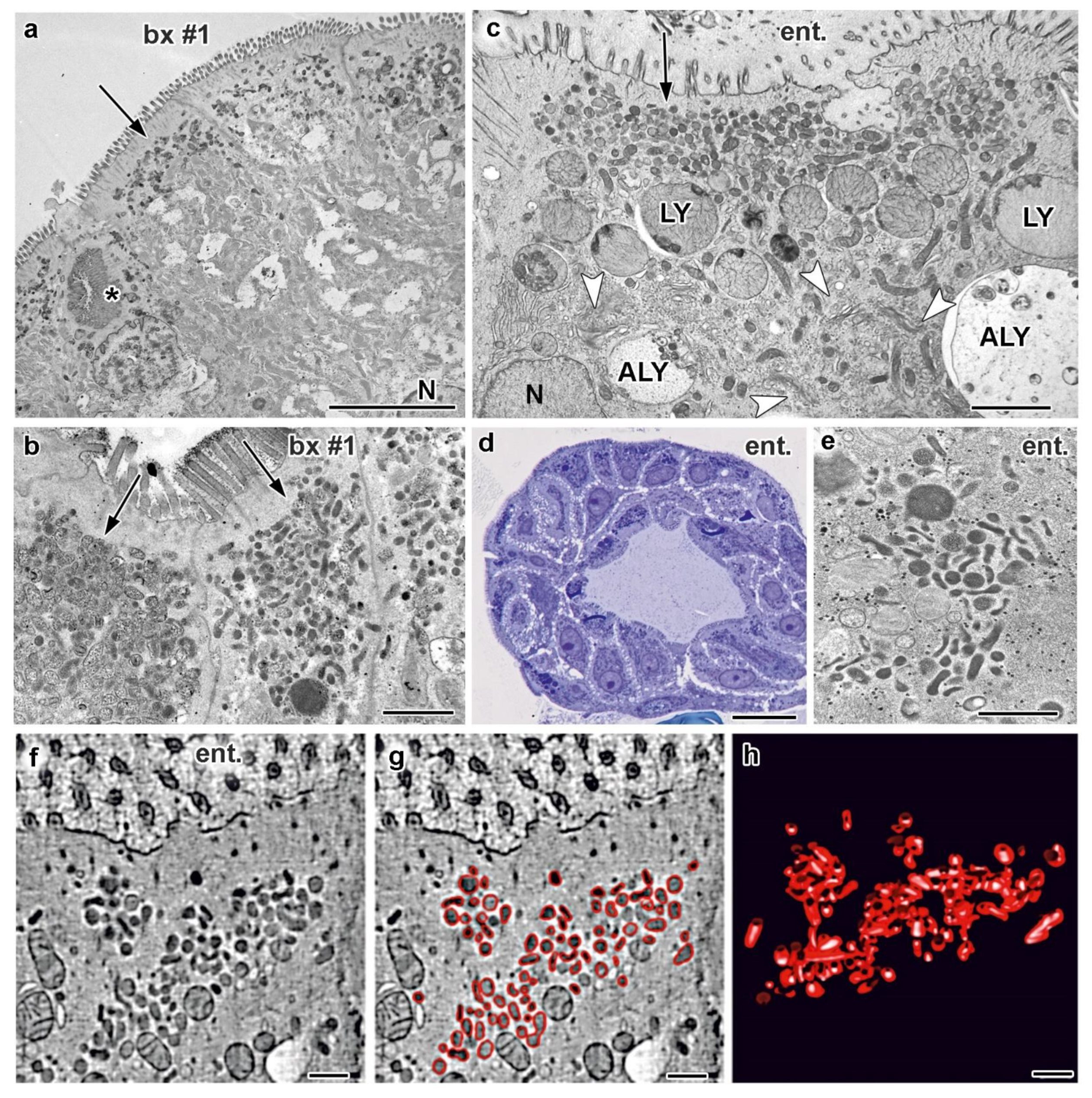
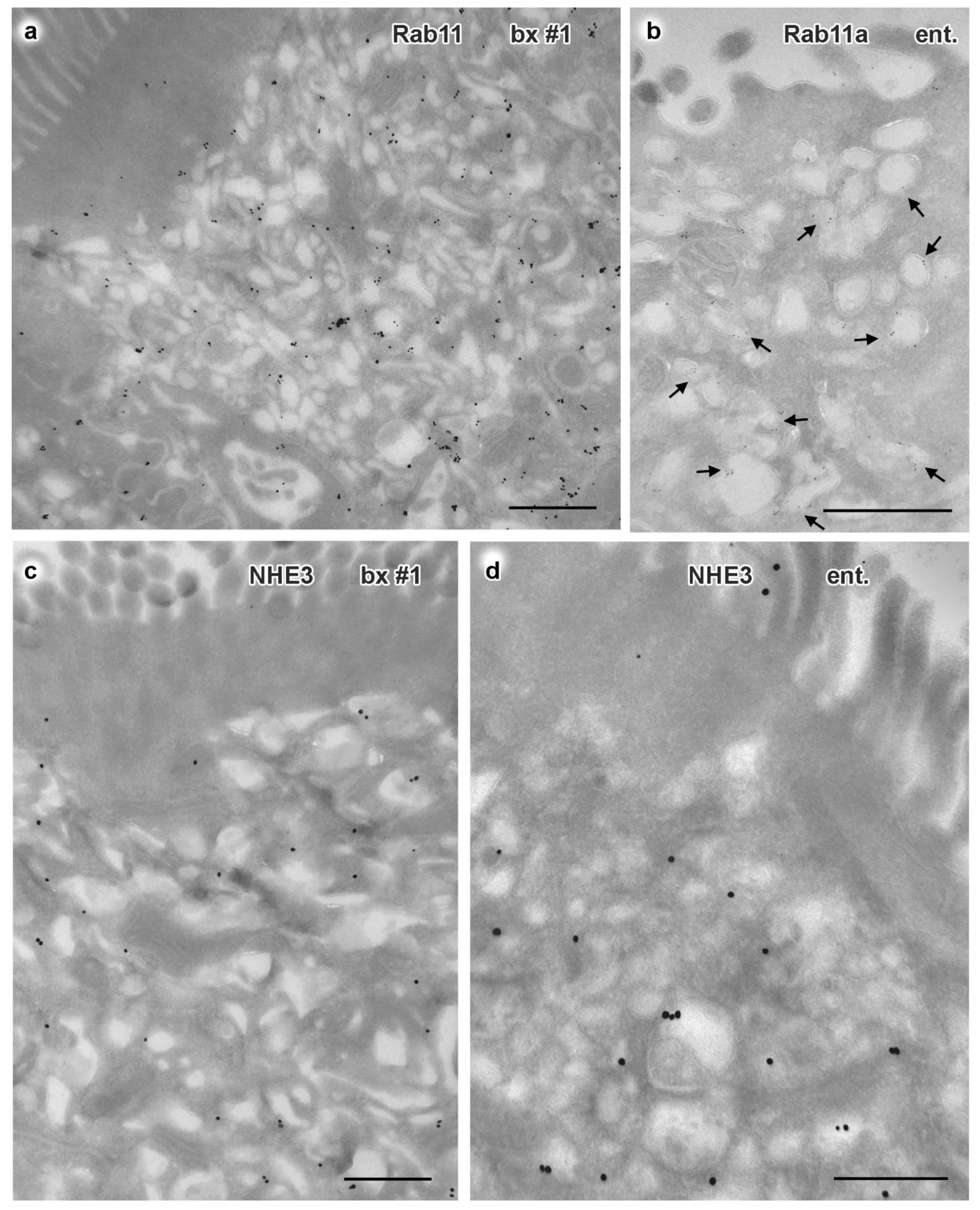
3.3. Electron Microscopy of Duodenal Biopsies and Enteroids
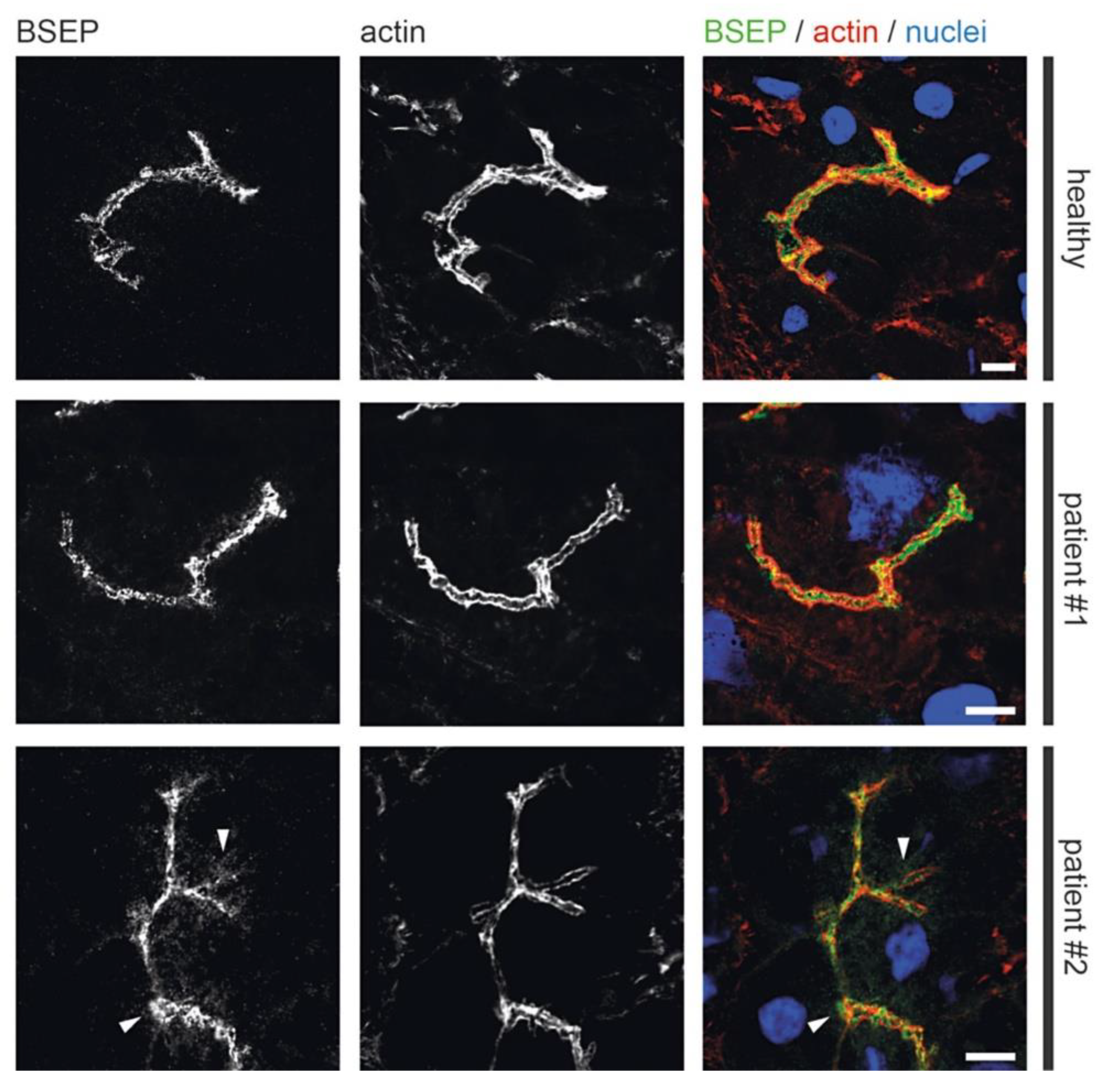
3.4. Fluorescence Microscopy of Liver Biopsies
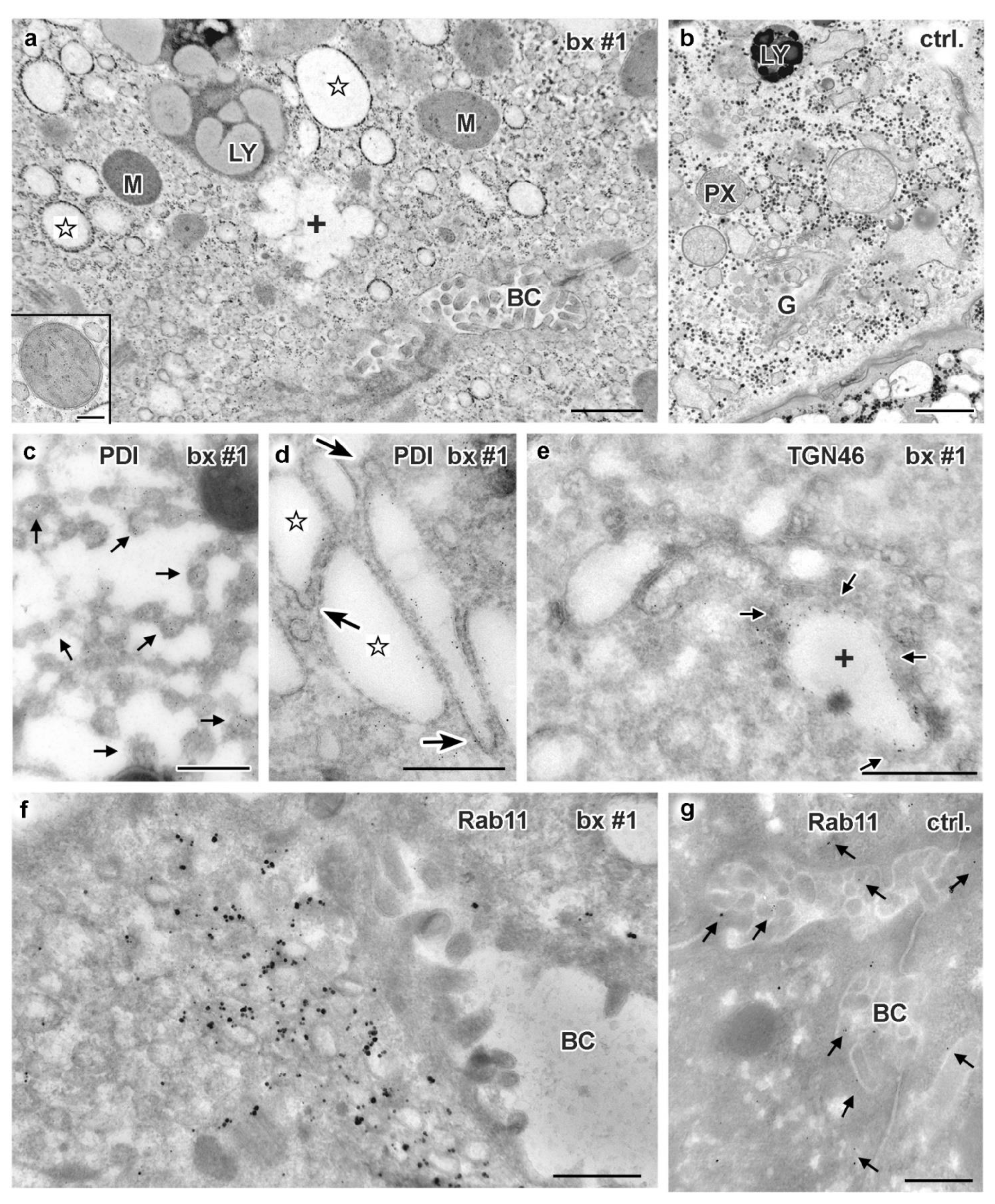
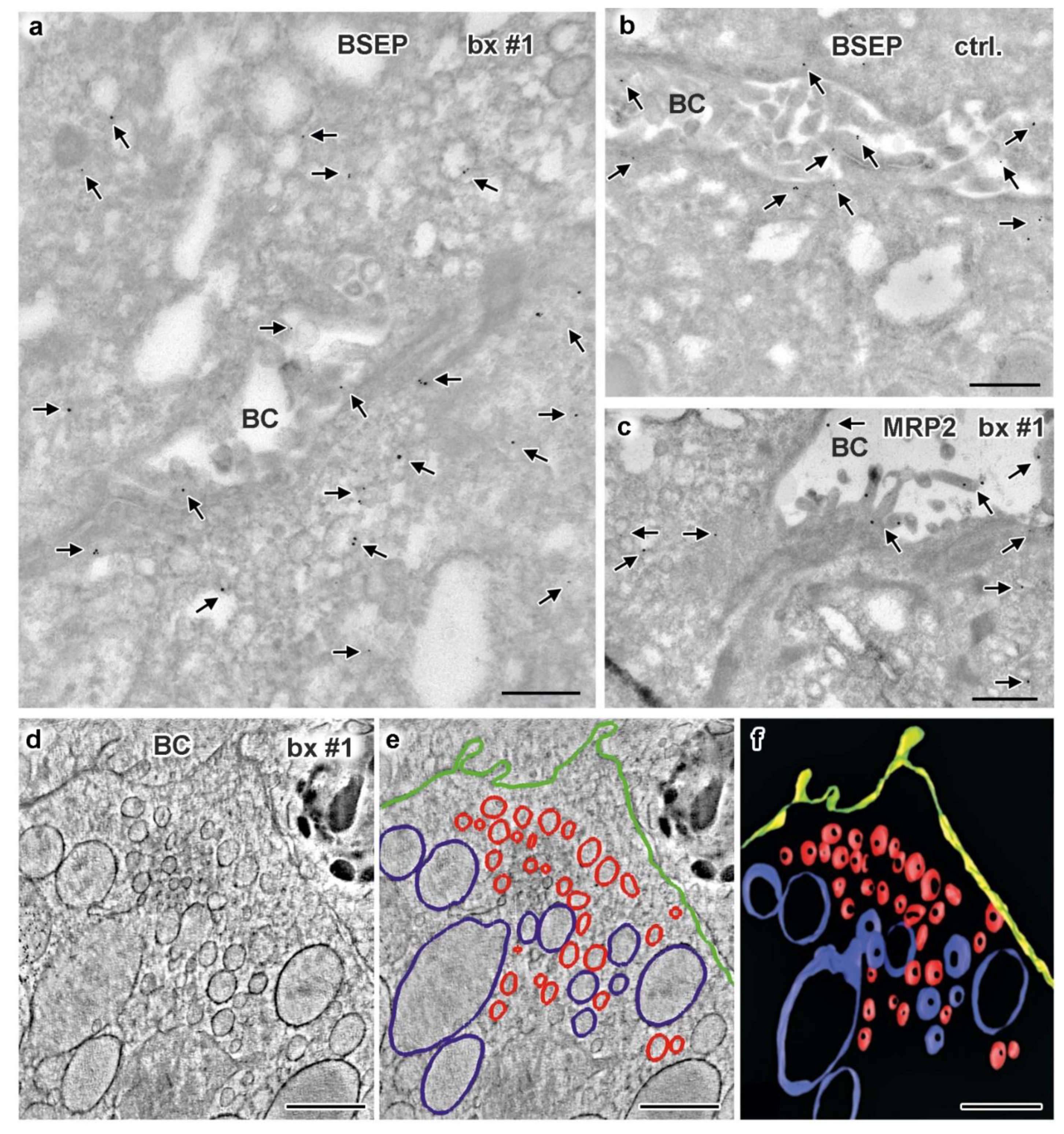
3.5. Electron Microscopy of Liver Biopsies
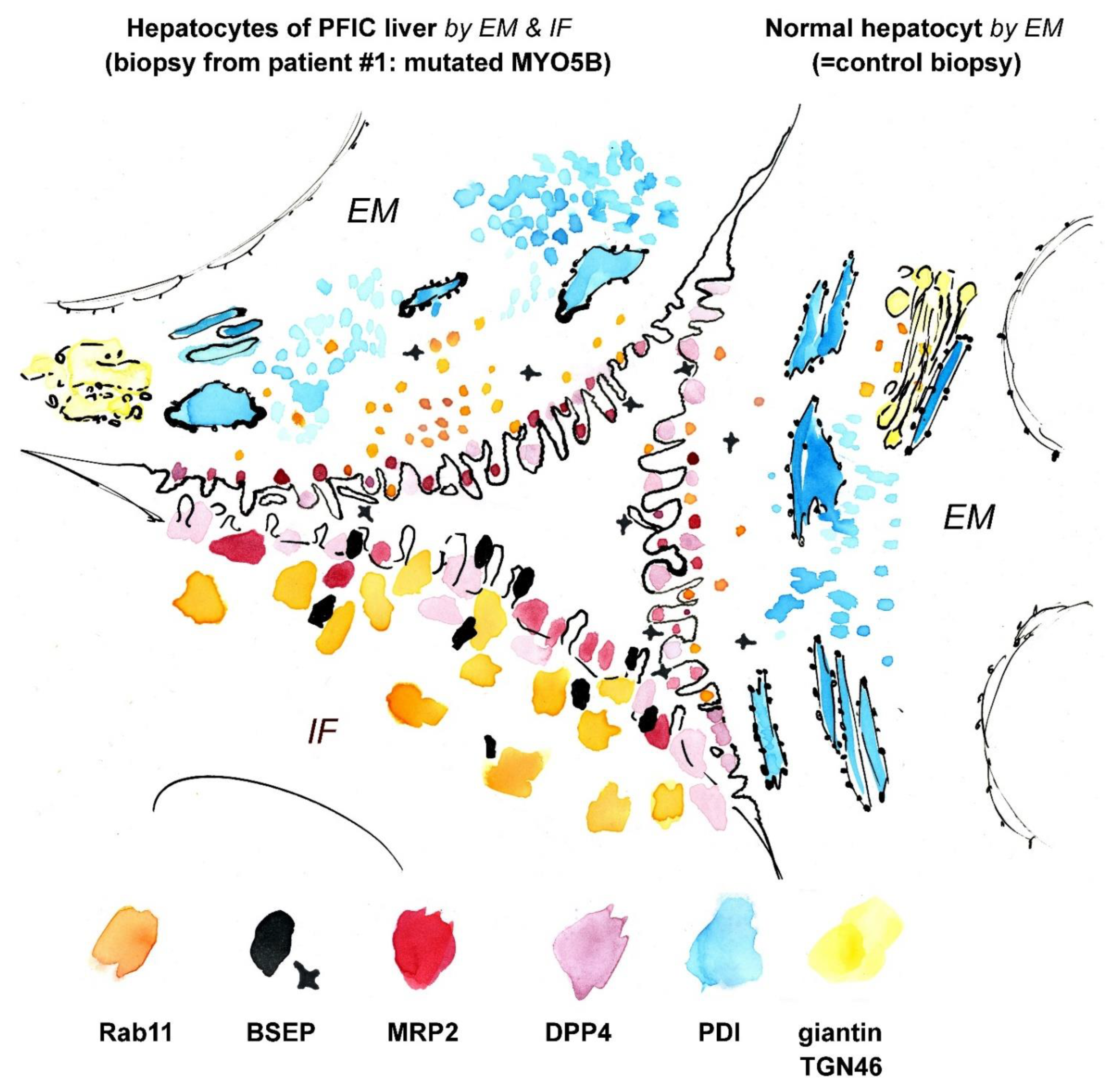
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qidwai, K.; Afkhami, M.; Day, C.E. The pathologist’s guide to fixatives. Methods Mol. Biol. 2014, 1180, 21–30. [Google Scholar] [PubMed]
- Geller, S.A. Liver: Tissue handling and evaluation. Methods Mol. Biol. 2014, 1180, 303–321. [Google Scholar]
- Ruemmele, F.M.; Müller, T.; Schiefermeier, N.; Ebner, H.L.; Lechner, S.; Pfaller, K.; Thöni, C.E.; Goulet, O.; Lacaille, F.; Schmitz, J.; et al. Loss-of-function of MYO5B is the main cause of microvillus inclusion disease: 15 novel mutations and a CaCo-2 RNAi cell model. Hum. Mutat. 2010, 31, 544–551. [Google Scholar] [CrossRef]
- Wiegerinck, C.L.; Janecke, A.R.; Schneeberger, K.; Vogel, G.F.; Van Haaften-Visser, D.Y.; Escher, J.C.; Adam, R.; Thöni, C.E.; Pfaller, K.; Jordan, A.J.; et al. Loss of Syntaxin 3 causes variant microvillus inclusion disease. Gastroenterology 2014, 147, 65–68.e10. [Google Scholar] [CrossRef]
- Vogel, G.F.; Klee, K.M.; Janecke, A.R.; Müller, T.; Hess, M.W.; Huber, L.A. Cargo-selective apical exocytosis in epithelial cells is conducted by Myo5B, Slp4a, Vamp7, and Syntaxin 3. J. Cell Biol. 2015, 211, 587–604. [Google Scholar] [CrossRef]
- Cartón-García, F.; Overeem, A.W.; Nieto, R.; Bazzocco, S.; Dopeso, H.; Macaya, I.; Bilic, J.; Landolfi, S.; Hernandez-Losa, J.; van Ijzendoorn, S.V.C.; et al. Myo5b knockout mice as a model of microvillus inclusion disease. Sci. Rep. 2015, 5, 12312. [Google Scholar] [CrossRef]
- Schneeberger, K.; Vogel, G.F.; Janecke, A.R.; Gerner, P.R.; Huber, L.A.; Hess, M.W.; Clevers, H.; Van Es, J.H.; Nieuwenhuis, E.E.S.; Middendorp, S.; et al. An inducible mouse model for microvillus inclusion disease reveals a role for myosin Vb in apical and basolateral trafficking. Proc. Natl. Acad. Sci. USA 2015, 112, 12408–12413. [Google Scholar] [CrossRef] [Green Version]
- Sidhaye, J.; Pinto, C.S.; Dharap, S.; Jacob, T.; Bhargava, S.; Sonawane, M. The zebrafish goosepimples/myosin Vb mutant exhibits cellular attributes of human microvillus inclusion disease. Mech. Dev. 2016, 142, 62–74. [Google Scholar] [CrossRef]
- Weis, V.G.; Knowles, B.C.; Choi, E.; Goldstein, A.E.; Williams, J.A.; Manning, E.H.; Roland, J.T.; Lapierre, L.A.; Goldenring, J.R. Loss of MYO5B in mice recapitulates microvillus inclusion disease and reveals an apical trafficking pathway distinct to neonatal duodenum. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 131–157. [Google Scholar] [CrossRef] [Green Version]
- Vogel, G.F.; Janecke, A.R.; Krainer, I.M.; Gutleben, K.; Witting, B.; Mitton, S.G.; Mansour, S.; Ballauff, A.; Roland, J.T.; Engevik, A.C.; et al. Abnormal Rab11-Rab8-vesicles cluster in enterocytes of patients with microvillus inclusion disease. Traffic 2017, 18, 453–464. [Google Scholar] [CrossRef]
- Vogel, G.F.; Van Rijn, J.M.; Krainer, I.M.; Janecke, A.R.; Posovzsky, C.; Cohen, M.; Searle, C.; Jantchou, P.; Escher, J.C.; Patey, N.; et al. Disrupted apical exocytosis of cargo vesicles causes enteropathy in FHL5 patients with Munc18-2 mutations. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Mosa, M.H.; Nicolle, O.; Maschalidi, S.; Sepulveda, F.E.; Bidaud-Meynard, A.; Menche, C.; Michels, B.E.; Michaux, G.; Basile, G.D.S.; Farin, H.F. Dynamic formation of microvillus inclusions during enterocyte differentiation in Munc18-2-deficient intestinal organoids. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 477–493.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidaud-Meynard, A.; Nicolle, O.; Heck, M.; Le Cunff, Y.; Michaux, G. A V0-ATPase-dependent apical trafficking pathway maintains the polarity of the intestinal absorptive membrane. Development 2019, 146, dev174508. [Google Scholar] [CrossRef] [Green Version]
- Overeem, A.W.; Klappe, K.; Parisi, S.; Klöters-Planchy, P.; Mataković, L.; du Teil Espina, M.; Drouin, C.A.; Weiss, K.H.; van IJzendoorn, S.C. Pluripotent stem cell-derived bile canaliculi-forming hepatocytes to study genetic liver diseases involving hepatocyte polarity. J. Hepatol. 2019, 71, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Engevik, A.C.; Coutts, A.W.; Kaji, I.; Rodriguez, P.; Ongaratto, F.; Saqui-Salces, M.; Medida, R.L.; Meyer, A.R.; Kolobova, E.; Engevik, M.A.; et al. Editing myosin VB gene to create porcine model of microvillus inclusion disease, with microvillus-lined inclusions and alterations in sodium transporters. Gastroenterology 2020, 158, 2236–2249.e9. [Google Scholar] [CrossRef] [PubMed]
- Jewett, C.E.; Appel, B.H.; Prekeris, R. The Rab11 effectors Fip5 and Fip1 regulate zebrafish intestinal development. Biol. Open 2020, 9, bio055822. [Google Scholar] [CrossRef]
- Müller, M.; Moor, H. Cryofixation of thick specimens by high-pressure freezing. In The Science of Biological Specimen Preparation; Revel, J.P., Barnard, T., Haggins, G.H., Eds.; SEM Inc. AMF O’Hare: Chicago, IL, USA, 1984; pp. 131–138. [Google Scholar]
- Humbel, B.; Müller, M. Freeze substitution and low temperature embedding. In The Science of Biological Specimen Preparation 1985; Müller, M., Becker, R.P., Wolosewick, J.J., Eds.; SEM Inc. AMF O’Hare: Chicago, IL, USA, 1986; pp. 175–183. [Google Scholar]
- Studer, D.; Michel, M.; Müller, M. High pressure freezing comes of age. Scanning Microsc. Suppl. 1989, 3, 253. [Google Scholar] [PubMed]
- Tokuyasu, K.T. A technique for ultracryotomy of cell suspensions and tissues. J. Cell Biol. 1973, 57, 551–565. [Google Scholar] [CrossRef]
- Liou, W.; Geuze, H.J.; Slot, J.W. Improving structural integrity of cryosections for immunogold labeling. Histochem. Cell Biol. 1996, 106, 41–58. [Google Scholar] [CrossRef]
- Liou, W.; Geuze, H.J.; Geelen, M.J.; Slot, J.W. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J. Cell Biol. 1997, 136, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Ameen, N.; Alexis, J.; Salas, P. Cellular localization of the cystic fibrosis transmembrane conductance regulator in mouse intestinal tract. Histochem. Cell Biol. 2000, 114, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Zeuschner, D.; Geerts, W.J.; Van Donselaar, E.; Humbel, B.M.; Slot, J.W.; Koster, A.J.; Klumperman, J. Immuno-electron tomography of ER exit sites reveals the existence of free COPII-coated transport carriers. Nat. Cell Biol. 2006, 8, 377–383. [Google Scholar] [CrossRef]
- Dombrowski, F.; Stieger, B.; Beuers, U. Tauroursodeoxycholic acid inserts the bile salt export pump into canalicular membranes of cholestatic rat liver. Lab. Investig. 2005, 86, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Kijanka, M.; Van Donselaar, E.; Müller, W.; Dorresteijn, B.; Popov-Čeleketić, D.; El Khattabi, M.; Verrips, C.; Henegouwen, P.V.B.E.; Post, J. A novel immuno-gold labeling protocol for nanobody-based detection of HER2 in breast cancer cells using immuno-electron microscopy. J. Struct. Biol. 2017, 199, 1–11. [Google Scholar] [CrossRef]
- Hess, M.W.; Vogel, G.F.; Yordanov, T.E.; Witting, B.; Gutleben, K.; Ebner, H.L.; De Araujo, M.E.G.; Filipek, P.A.; Huber, L.A. Combining high-pressure freezing with pre-embedding immunogold electron microscopy and tomography. Traffic 2018, 19, 639–649. [Google Scholar] [CrossRef]
- Davidson, G.P.; Cutz, E.; Hamilton, J.R.; Gall, D.G. Familial enteropathy: A syndrome of protracted diarrhea from birth, failure to thrive, and hypoplastic villus atrophy. Gastroenterology 1978, 75, 783–790. [Google Scholar] [CrossRef]
- Cutz, E.; Rhoads, J.M.; Drumm, B.; Sherman, P.M.; Durie, P.R.; Forstner, G.G. Microvillus inclusion disease: An inherited defect of brush-border assembly and differentiation. N. Engl. J. Med. 1989, 320, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.D.; Schmitz, J. Familial microvillous atrophy: A clinicopathological survey of 23 cases. J. Pediatr. Gastroenterol. Nutr. 1992, 14, 380–396. [Google Scholar] [CrossRef]
- Iancu, T.C.; Mahajnah, M.; Manov, I.; Shaoul, R. Microvillous inclusion disease: Ultrastructural variability. Ultrastruct. Pathol. 2007, 31, 173–188. [Google Scholar] [CrossRef]
- Ruemmele, F.M.; Schmitz, J.; Goulet, O. Microvillous inclusion disease (microvillous atrophy). Orphanet J. Rare Dis. 2006, 1, 22. [Google Scholar] [CrossRef] [Green Version]
- Jayawardena, D.; Alrefai, W.A.; Dudeja, P.K.; Gill, R.K. Recent advances in understanding and managing malabsorption: Focus on microvillus inclusion disease. F1000Research 2019, 8, 2061. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.; Hess, M.W.; Schiefermeier, N.; Pfaller, K.; Ebner, H.L.; Heinz-Erian, P.; Ponstingl, H.; Partsch, J.; Röllinghoff, B.; Köhler, H.; et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat. Genet. 2008, 40, 1163–1165. [Google Scholar] [CrossRef]
- Phillips, A.D.; Szafranski, M.; Man, L.-Y.; Wall, W.J. Periodic acid-schiff staining abnormality in microvillous atrophy: Photometric and ultrastructural studies. J. Pediatr. Gastroenterol. Nutr. 2000, 30, 34–42. [Google Scholar] [CrossRef]
- Sherman, P.M.; Mitchell, D.J.; Cutz, E. Neonatal enteropathies: Defining the causes of protracted diarrhea of infancy. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, C.; Weis, V.G.; Knowles, B.C.; Lapierre, L.A.; Martin, M.G.; Dickman, P.; Goldenring, J.R.; Shub, M.D. Apical membrane alterations in non-intestinal organs in microvillus inclusion disease. Dig. Dis. Sci. 2018, 63, 356–365. [Google Scholar] [CrossRef]
- Girard, M.; Lacaille, F.; Verkarre, V.; Mategot, R.; Feldmann, G.; Grodet, A.; Sauvat, F.; Irtan, S.; Davit-Spraul, A.; Jacquemin, E.; et al. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology 2014, 60, 301–310. [Google Scholar] [CrossRef]
- Qiu, Y.-L.; Gong, J.-Y.; Wang, N.-L.; Yan, Y.-Y.; Li, J.-Q.; Chen, L.; Borchers, C.H.; Sipos, B.; Knisely, A.S.; Ling, V.; et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low & gamma-glutamyltransferase cholestasis. Hepatology 2017, 65, 1655–1669. [Google Scholar] [CrossRef]
- Bull, L.N.; Thompson, R.J. Progressive familial intrahepatic cholestasis. Clin. Liver Dis. 2018, 22, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Overeem, A.W.; Li, Q.; Van Ijzendoorn, S.C.; Qiu, Y.; Cartón--García, F.; Leng, C.; Klappe, K.; Dronkers, J.; Hsiao, N.; Wang, J.; et al. A molecular mechanism underlying genotype--specific intrahepatic cholestasis resulting from MYO5B mutations. Hepatology 2019, 72, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, E.; Taylor, S.A.; Davit--Spraul, A.; Thébaut, A.; Thomassin, N.; Guettier, C.; Whitington, P.F.; Jacquemin, E. MYO5B mutations cause cholestasis with normal serum gamma—glutamyl transferase activity in children without microvillous inclusion disease. Hepatology 2017, 65, 164–173. [Google Scholar] [CrossRef]
- Van Ijzendoorn, S.C.D.; Li, Q.; Qiu, Y.L.; Wang, J.S.; Overeem, A.W. Unequal effects of myosin 5B mutations in liver and intestine determine the clinical presentation of low-gamma-glutamyltransferase cholestasis. Hepatology 2020, 72, 1461–1468. [Google Scholar] [CrossRef] [PubMed]
- Aldrian, D.; Vogel, G.; Frey, T.; Civan, H.A.; Aksu, A.; Avitzur, Y.; Ramos, E.B.; Çakır, M.; Demir, A.; Deppisch, C.; et al. Congenital diarrhea and cholestatic liver disease: Phenotypic spectrum associated with MYO5B mutations. J. Clin. Med. 2021, 10, 481. [Google Scholar] [CrossRef]
- Griffiths, G. Fine Structure Immunocytochemistry; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1993. [Google Scholar]
- Webster, P.; Webster, A. Cryosectioning fixed and cryoprotected biological material for immunocytochemistry. Methods Mol. Biol. 2014, 1117, 273–313. [Google Scholar]
- Hess, M.W.; Pfaller, K.; Ebner, H.L.; Beer, B.; Hekl, D.; Seppi, T. 3D versus 2D cell culture implications for electron microscopy. Methods Cell Biol. 2010, 96, 649–670. [Google Scholar] [PubMed]
- Walther, P.; Ziegler, A. Freeze substitution of high-pressure frozen samples: The visibility of biological membranes is improved when the substitution medium contains water. J. Microsc. 2002, 208, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiéry, J.P. Mise en évidence des polysaccharides sur coupes fines en microscopie électronique. J. Microsc. 1967, 6, 987–1018. (In French) [Google Scholar]
- Neiss, W.F. Enhancement of the periodic acid-Schiff (PAS) and periodic acid-thiocarbohydrazide-silver proteinate (PA-TCH-SP) reaction in LR white sections. Histochemistry 1988, 88, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Berod, A.; Hartman, B.K.; Pujol, J.F. Importance of fixation in immunohistochemistry: Use of formaldehyde solutions at variable pH for the localization of tyrosine hydroxylase. J. Histochem. Cytochem. 1981, 29, 844–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapierre, L.A.; Ducharme, N.A.; Drake, K.R.; Goldenring, J.R.; Kenworthy, A.K. Coordinated regulation of caveolin-1 and Rab11a in apical recycling compartments of polarized epithelial cells. Exp. Cell Res. 2012, 318, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Roland, J.T.; Kenworthy, A.K.; Peranen, J.; Caplan, S.; Goldenring, J.R. Myosin Vb interacts with Rab8a on a tubular network containing EHD1 and EHD3. Mol. Biol. Cell 2007, 18, 2828–2837. [Google Scholar] [CrossRef] [Green Version]
- Filimonenko, M.; Stuffers, S.; Raiborg, C.; Yamamoto, A.; Malerød, L.; Fisher, E.M.; Isaacs, A.; Brech, A.; Stenmark, H.; Simonsen, A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 2007, 179, 485–500. [Google Scholar] [CrossRef]
- Vogel, G.F.; Ebner, H.L.; De Araujo, M.E.G.; Schmiedinger, T.; Eiter, O.; Pircher, H.; Gutleben, K.; Witting, B.; Teis, D.; Huber, L.A.; et al. Ultrastructural morphometry points to a new role for LAMTOR2 in regulating the endo/lysosomal system. Traffic 2015, 16, 617–634. [Google Scholar] [CrossRef]
- Pizzo, L.; Fariello, M.I.; Lepanto, P.; Aguilar, P.S.; Kierbel, A. An image analysis method to quantify CFTR subcellular localization. Mol. Cell. Probes 2014, 28, 175–180. [Google Scholar] [CrossRef]
- Cui, L.; Aleksandrov, L.; Chang, X.-B.; Hou, Y.-X.; He, L.; Hegedus, T.; Gentzsch, M.; Aleksandrov, A.; Balch, W.E.; Riordan, J.R. Domain interdependence in the biosynthetic assembly of CFTR. J. Mol. Biol. 2007, 365, 981–994. [Google Scholar] [CrossRef]
- Kromeyer-Hauschild, K.; Wabitsch, M.; Kunze, D.; Geller, F.; Geisz, H.C.; Hesse, V.; Von Hippel, A.; Jaeger, U.; Johnsen, D.; Korte, W.; et al. Percentiles of body mass index in children and adolescents evaluated from different regional German studies. Monatsschr. Kinderh. 2001, 149, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Goldenring, J.R. The challenge of personalized cell biology: The example of microvillus inclusion disease. Traffic 2020, 21, 169–171. [Google Scholar] [CrossRef]
- Murk, J.L.A.N.; Posthuma, G.; Koster, A.J.; Geuze, H.J.; Verkleij, A.J.; Kleijmeer, M.J.; Humbel, B.M. Influence of aldehyde fixation on the morphology of endosomes and lysosomes: Quantitative analysis and electron tomography. J. Microsc. 2003, 212, 81–90. [Google Scholar] [CrossRef]
- Ladinsky, M.S.; Mastronarde, D.N.; McIntosh, J.R.; Howell, K.E.; Staehelin, L.A. Golgi structure in three dimensions: Functional insights from the normal rat kidney cell. J. Cell Biol. 1999, 144, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- Hess, M.W.; Müller, M.; Debbage, P.L.; Vetterlein, M.; Pavelka, M. Cryopreparation provides new insight into the effects of brefeldin A on the structure of the HepG2 golgi apparatus. J. Struct. Biol. 2000, 130, 63–72. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Ladinsky, M.S.; Huey-Tubman, K.E.; Jensen, G.J.; McIntosh, J.R.; Bjorkman, P.J. FcRn-mediated antibody transport across epithelial cells revealed by electron tomography. Nature 2008, 455, 542–546. [Google Scholar] [CrossRef]
- Overeem, A.; Posovszky, C.; Rings, E.H.M.M.; Giepmans, B.; Van Ijzendoorn, S. The role of enterocyte defects in the pathogenesis of congenital diarrheal disorders. Dis. Model. Mech. 2016, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, R.; Nicastro, D.; Mastronarde, D. New views of cells in 3D: An introduction to electron tomography. Trends Cell Biol. 2005, 15, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.R.; Gibson, A.; Shipman, M.; Miller, K. Movement of internalized ligand-receptor complexes along a continuous endosomal reticulum. Nat. Cell Biol. 1990, 346, 335–339. [Google Scholar] [CrossRef]
- Troughton, W.D.; Trier, J.S. Paneth and goblet cell renewal in mouse duodenal crypts. J. Cell Biol. 1969, 41, 251–268. [Google Scholar] [CrossRef]
- Kaji, I.; Roland, J.T.; Watanabe, M.; Engevik, A.C.; Goldstein, A.E.; Hodges, C.A.; Goldenring, J.R. Lysophosphatidic acid increases maturation of brush borders and SGLT1 activity in MYO5B-deficient mice, a model of microvillus inclusion disease. Gastroenterology 2020, 159, 1390–1405.e20. [Google Scholar] [CrossRef]
- Leng, C.; Overeem, A.W.; Cartón-Garcia, F.; Li, Q.; Klappe, K.; Kuipers, J.; Cui, Y.; Zuhorn, I.S.; Arango, D.; Van Ijzendoorn, S.C.D. Loss of MYO5B expression deregulates late endosome size which hinders mitotic spindle orientation. PLoS Biol. 2019, 17, e3000531. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, E. Lysosomes; Plenum Press: New York, NY, USA, 1989. [Google Scholar]
- Fengsrud, M.; Roos, N.; Berg, T.; Liou, W.; Slot, J.W.; Seglen, P.O. Ultrastructural and immunocytochemical characterization of autophagic vacuoles in isolated hepatocytes: Effects of vinblastine and asparagine on vacuole distributions. Exp. Cell Res. 1995, 221, 504–519. [Google Scholar] [CrossRef]
- Reinshagen, K.; Naim, H.Y.; Zimmer, K.-P. Autophagocytosis of the apical membrane in microvillus inclusion disease. Gut 2002, 51, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Ameen, N.A.; Van Donselaar, E.; Posthuma, G.; De Jonge, H.; McLaughlin, G.; Geuze, H.J.; Marino, C.; Peters, P.J. Subcellular distribution of CFTR in rat intestine supports a physiologic role for CFTR regulation by vesicle traffic. Histochem. Cell Biol. 2000, 114, 219–228. [Google Scholar] [CrossRef]
- Engevik, A.C.; Kaji, I.; Engevik, M.A.; Meyer, A.R.; Weis, V.G.; Goldstein, A.; Hess, M.W.; Müller, T.; Koepsell, H.; Dudeja, P.K.; et al. Loss of MYO5B leads to reductions in Na+ absorption with maintenance of CFTR-dependent Cl-secretion in enterocytes. Gastroenterology 2018, 155, 1883–1897.e10. [Google Scholar] [CrossRef] [PubMed]
- Knowles, B.C.; Roland, J.T.; Krishnan, M.; Tyska, M.J.; Lapierre, L.A.; Dickman, P.S.; Goldenring, J.R.; Shub, M.D. Myosin Vb uncoupling from RAB8A and RAB11A elicits microvillus inclusion disease. J. Clin. Investig. 2014, 124, 2947–2962. [Google Scholar] [CrossRef] [Green Version]
- Steiner, J.W.; Carruthers, J.S.; Kalifat, S.R. Observations on the fine structure of rat liver cells in extrahepatic cholestasis. Z. Zellforsch. Mikrosk Anat. 1962, 58, 141–159. [Google Scholar] [CrossRef]
- Jones, A.L.; Fawcett, D.W. Hypertrophy of the agranular endoplasmic reticulum in hamster liver induced by phenobarbital (with a review on the functions of this organelle in liver). J. Histochem. Cytochem. 1966, 14, 215–232. [Google Scholar] [CrossRef]
- Ghadially, F.N. Ultrastructural Pathology of the Cell and Matrix; Butterworth-Heinemann: Oxford, UK, 1997. [Google Scholar]
- Sakaguchi, T.; Leser, G.P.; Lamb, R.A. The ion channel activity of the influenza virus M2 protein affects transport through the Golgi apparatus. J. Cell Biol. 1996, 133, 733–747. [Google Scholar] [CrossRef]
- Mori, Y.; Koike, M.; Moriishi, E.; Kawabata, A.; Tang, H.; Oyaizu, H.; Uchiyama, Y.; Yamanishi, K. Human herpesvirus-6 induces MVB formation, and virus egress occurs by an exosomal release pathway. Traffic 2008, 9, 1728–1742. [Google Scholar] [CrossRef] [Green Version]
- Phillips, A.; Fransen, J.; Hauri, H.-P.; Sterchi, E. The constitutive exocytotic pathway in microvillous atrophy. J. Pediatr. Gastroenterol. Nutr. 1993, 17, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Hunziker, E.B.; Herrmann, W.; Schenk, R.K.; Mueller, M.; Moor, H. Cartilage ultrastructure after high pressure freezing, freeze substitution, and low temperature embedding. I. Chondrocyte ultrastructure—implications for the theories of mineralization and vascular invasion. J. Cell Biol. 1984, 98, 267–276. [Google Scholar] [CrossRef]
- Szczesny, P.J.; Walther, P.; Müller, M. Light damage in rod outer segments: The effects of fixation on ultrastructural alterations. Curr. Eye Res. 1996, 15, 807–814. [Google Scholar] [CrossRef]
- Pfeiffer, S.; Vielhaber, G.; Vietzke, J.-P.; Wittern, K.-P.; Hintze, U.; Wepf, R. High-pressure freezing provides new information on human epidermis: Simultaneous protein antigen and lamellar lipid structure preservation. study on human epidermis by cryoimmobilization. J. Investig. Dermatol. 2000, 114, 1030–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, M.W. Cryopreparation methodology for plant cell biology. Methods Cell Biol. 2007, 79, 57–100. [Google Scholar]
- Ameen, N.A.; Salas, P.J. Microvillus inclusion disease: A genetic defect affecting apical membrane protein traffic in intestinal epithelium. Traffic 2000, 1, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Van Rijn, J.M.; Ardy, R.C.; Kuloğlu, Z.; Härter, B.; van Haaften-Visser, D.Y.; van der Doef, H.P.; van Hoesel, M.; Kansu, A.; van Vugt, A.H.; Thian, M.; et al. Intestinal failure and aberrant lipid metabolism in patients with DGAT1 deficiency. Gastroenterology 2018, 155, 130–143.e15. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, D.D.; Bensch, K.; Barrnett, R.J. Cytochemistry and electron microscopy. The preservation of cellular ultrastructure and enzymatic activity by aldehyde fixation. J. Cell Biol. 1963, 17, 19–58. [Google Scholar] [CrossRef]
- Möbius, W.; Posthuma, G. Sugar and ice: Immunoelectron microscopy using cryosections according to the Tokuyasu method. Tissue Cell 2019, 57, 90–102. [Google Scholar] [CrossRef]
- Solberg, L.B.; Melhus, G.; Brorson, S.-H.; Wendel, M.; Reinholt, F.P. Heat-induced retrieval of immunogold labeling for nucleobindin and osteoadherin from Lowicryl sections of bone. Micron 2006, 37, 347–354. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hess, M.W.; Krainer, I.M.; Filipek, P.A.; Witting, B.; Gutleben, K.; Vietor, I.; Zoller, H.; Aldrian, D.; Sturm, E.; Goldenring, J.R.; et al. Advanced Microscopy for Liver and Gut Ultrastructural Pathology in Patients with MVID and PFIC Caused by MYO5B Mutations. J. Clin. Med. 2021, 10, 1901. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10091901
Hess MW, Krainer IM, Filipek PA, Witting B, Gutleben K, Vietor I, Zoller H, Aldrian D, Sturm E, Goldenring JR, et al. Advanced Microscopy for Liver and Gut Ultrastructural Pathology in Patients with MVID and PFIC Caused by MYO5B Mutations. Journal of Clinical Medicine. 2021; 10(9):1901. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10091901
Chicago/Turabian StyleHess, Michael W., Iris M. Krainer, Przemyslaw A. Filipek, Barbara Witting, Karin Gutleben, Ilja Vietor, Heinz Zoller, Denise Aldrian, Ekkehard Sturm, James R. Goldenring, and et al. 2021. "Advanced Microscopy for Liver and Gut Ultrastructural Pathology in Patients with MVID and PFIC Caused by MYO5B Mutations" Journal of Clinical Medicine 10, no. 9: 1901. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10091901