
Pharmacologic Management for Ventricular Arrhythmias: Overview of Anti-Arrhythmic Drugs


Abstract
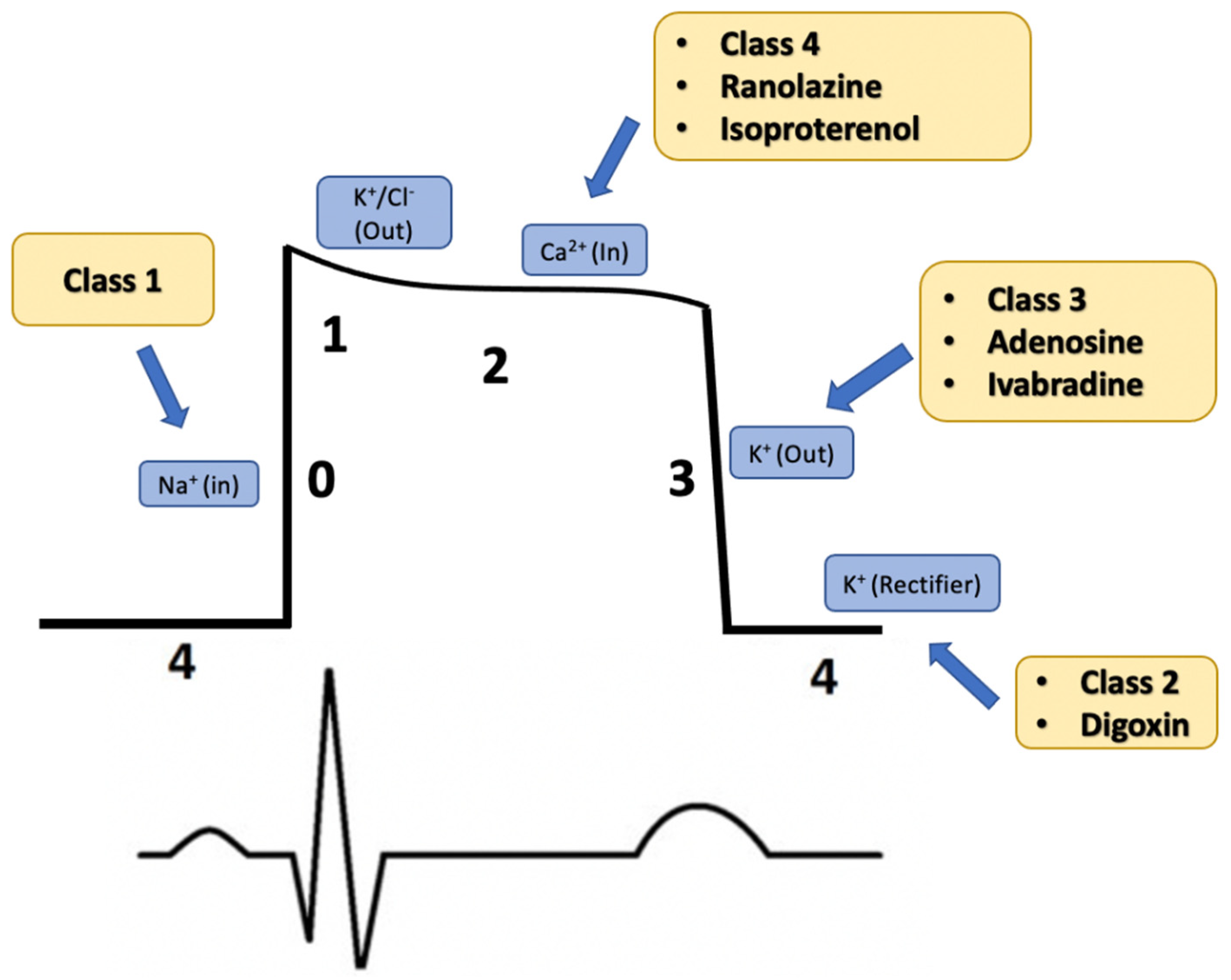
:1. Introduction
2. Drug Classification
3. Class I Medications: Sodium Channel Blockade
3.1. Class IA
3.1.1. Procainamide
3.1.2. Quinidine
3.1.3. Disopyramide
3.2. Class IB
3.2.1. Lidocaine
3.2.2. Mexiletine
3.3. Class IC
Flecainide and Propafenone
4. Class II Medications: Beta Blockade
5. Class III Medications: Potassium Channel Blockade
5.1. Amiodarone
5.2. Dronedarone
5.3. Dofetilide
5.4. Sotalol
6. Class IV Medications: Calcium Channel Blockade
7. Medications Outside of the Classification System
7.1. Ranolazine
7.2. Adenosine
7.3. Digoxin
7.4. Isoproterenol
7.5. Ivabradine
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 2018, 138, e210–e271. [Google Scholar] [CrossRef] [Green Version]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proietti, R.; Labos, C.; Davis, M.; Thanassoulis, G.; Santangeli, P.; Russo, V.; Di Biase, L.; Roux, J.F.; Verma, A.; Natale, A.; et al. A systematic review and meta-analysis of the association between implantable cardioverter-defibrillator shocks and long-term mortality. Can. J. Cardiol. 2015, 31, 270–277. [Google Scholar] [CrossRef]
- Germano, J.J.; Reynolds, M.; Essebag, V.; Josephson, M.E. Frequency and causes of implantable cardioverter-defibrillator therapies: Is device therapy proarrhythmic? Am. J. Cardiol. 2006, 97, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Antiarrhythmics versus Implantable Defibrillators (AVID) Investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N. Engl. J. Med. 1997, 337, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- van Welsenes, G.H.; van Rees, J.B.; Borleffs, C.J.; Cannegieter, S.C.; Bax, J.J.; van Erven, L.; Schalij, M.J. Long-term follow-up of primary and secondary prevention implantable cardioverter defibrillator patients. Europace 2011, 13, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Santangeli, P.; Muser, D.; Maeda, S.; Filtz, A.; Zado, E.S.; Frankel, D.S.; Dixit, S.; Epstein, A.E.; Callans, D.J.; Marchlinski, F.E. Comparative effectiveness of antiarrhythmic drugs and catheter ablation for the prevention of recurrent ventricular tachycardia in patients with implantable cardioverter-defibrillators: A systematic review and meta-analysis of randomized controlled trials. Heart Rhythm 2016, 13, 1552–1559. [Google Scholar] [CrossRef] [PubMed]
- AlTurki, A.; Proietti, R.; Russo, V.; Dhanjal, T.; Banerjee, P.; Essebag, V. Anti-arrhythmic drug therapy in implantable cardioverter-defibrillator recipients. Pharmacol. Res. 2019, 143, 133–142. [Google Scholar] [CrossRef]
- Vaughan Williams, E.M. Significance of classifying antiarrhythmic actions since the cardiac arrhythmia suppression trial. J. Clin. Pharmacol. 1991, 31, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Wu, L.; Terrar, D.A.; Huang, C.L. Modernized Classification of Cardiac Antiarrhythmic Drugs. Circulation 2018, 138, 1879–1896. [Google Scholar] [CrossRef]
- Kidwell, G.A.; Greenspon, A.J.; Greenberg, R.M.; Volosin, K.J. Use-dependent prolongation of ventricular tachycardia cycle length by type I antiarrhythmic drugs in humans. Circulation 1993, 87, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Grace, A.A.; Camm, A.J. Quinidine. N. Engl. J. Med. 1998, 338, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Giardina, E.-G.V. Procainamide: Clinical Pharmacology and Efficacy against Ventricular Arrhythmiasa. Ann. N. Y. Acad. Sci. 1984, 432, 177–188. [Google Scholar] [CrossRef]
- Koch-Weser, J. Disopyramide. N. Engl. J. Med. 1979, 300, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Marill, K.A.; deSouza, I.S.; Nishijima, D.K.; Senecal, E.L.; Setnik, G.S.; Stair, T.O.; Ruskin, J.N.; Ellinor, P.T. Amiodarone or procainamide for the termination of sustained stable ventricular tachycardia: An historical multicenter comparison. Acad. Emerg. Med. 2010, 17, 297–306. [Google Scholar] [CrossRef]
- Gorgels, A.P.; van den Dool, A.; Hofs, A.; Mulleneers, R.; Smeets, J.L.; Vos, M.A.; Wellens, H.J. Comparison of procainamide and lidocaine in terminating sustained monomorphic ventricular tachycardia. Am. J. Cardiol. 1996, 78, 43–46. [Google Scholar] [CrossRef]
- Ortiz, M.; Martín, A.; Arribas, F.; Coll-Vinent, B.; Del Arco, C.; Peinado, R.; Almendral, J.; Investigators, P.S. Randomized comparison of intravenous procainamide vs. intravenous amiodarone for the acute treatment of tolerated wide QRS tachycardia: The PROCAMIO study. Eur. Heart J. 2017, 38, 1329–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, J.W. A Comparison of Seven Antiarrhythmic Drugs in Patients with Ventricular Tachyarrhythmias. N. Engl. J. Med. 1993, 329, 452–458. [Google Scholar] [CrossRef]
- Toniolo, M.; Muser, D.; Grilli, G.; Burelli, M.; Rebellato, L.; Daleffe, E.; Facchin, D.; Imazio, M. Oral procainamide as pharmacological treatment of recurrent and refractory ventricular tachyarrhythmias: A single-center experience. Heart Rhythm O2 2021, 2, 840–847. [Google Scholar] [CrossRef]
- Horowitz, L.N.; Josephson, M.E.; Farshidi, A.; Spielman, S.R.; Michelson, E.L.; Greenspan, A.M. Recurrent sustained ventricular tachycardia 3. Role of the electrophysiologic study in selection of antiarrhythmic regimens. Circulation 1978, 58, 986–997. [Google Scholar] [CrossRef] [Green Version]
- Myerburg, R.J.; Kessler, K.M.; Kiem, I.; Pefkaros, K.C.; Conde, C.A.; Cooper, D.; Castellanos, A. Relationship between plasma levels of procainamide, suppression of premature ventricular complexes and prevention of recurrent ventricular tachycardia. Circulation 1981, 64, 280–290. [Google Scholar] [CrossRef] [Green Version]
- Brugada, R.; Brugada, J.; Antzelevitch, C.; Kirsch, G.E.; Potenza, D.; Towbin, J.A.; Brugada, P. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation 2000, 101, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Haïssaguerre, M.; Sacher, F.; Nogami, A.; Komiya, N.; Bernard, A.; Probst, V.; Yli-Mayry, S.; Defaye, P.; Aizawa, Y.; Frank, R.; et al. Characteristics of recurrent ventricular fibrillation associated with inferolateral early repolarization role of drug therapy. J. Am. Coll. Cardiol. 2009, 53, 612–619. [Google Scholar] [CrossRef] [Green Version]
- Viskin, S.; Chorin, E.; Viskin, D.; Hochstadt, A.; Halkin, A.; Tovia-Brodie, O.; Lee, J.K.; Asher, E.; Laish-Farkash, A.; Amit, G.; et al. Quinidine-Responsive Polymorphic Ventricular Tachycardia in Patients With Coronary Heart Disease. Circulation 2019, 139, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Viskin, S.; Hochstadt, A.; Chorin, E.; Viskin, D.; Havakuk, O.; Khoury, S.; Lee, J.K.; Belhassen, B.; Rosso, R. Quinidine-responsive out-of-hospital polymorphic ventricular tachycardia in patients with coronary heart disease. Europace 2020, 22, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Larson, J.; Ghannam, M.; Saeed, M.; Cunnane, R.; Ghanbari, H.; Latchamsetty, R.; Crawford, T.; Jongnarangsin, K.; Pelosi, F.; et al. Efficacy and tolerability of quinidine as salvage therapy for monomorphic ventricular tachycardia in patients with structural heart disease. J. Cardiovasc. Electrophysiol. 2021, 32, 3173–3178. [Google Scholar] [CrossRef] [PubMed]
- Morganroth, J.; Goin, J.E. Quinidine-related mortality in the short-to-medium-term treatment of ventricular arrhythmias. A meta-analysis. Circulation 1991, 84, 1977–1983. [Google Scholar] [CrossRef] [Green Version]
- Li, D.L.; Cox, Z.L.; Richardson, T.D.; Kanagasundram, A.N.; Saavedra, P.J.; Shen, S.T.; Montgomery, J.A.; Murray, K.T.; Roden, D.M.; Stevenson, W.G. Quinidine in the Management of Recurrent Ventricular Arrhythmias: A Reappraisal. JACC Clin. Electrophysiol. 2021, 7, 1254–1263. [Google Scholar] [CrossRef]
- Hermida, J.S.; Denjoy, I.; Clerc, J.; Extramiana, F.; Jarry, G.; Milliez, P.; Guicheney, P.; Di Fusco, S.; Rey, J.L.; Cauchemez, B.; et al. Hydroquinidine therapy in Brugada syndrome. J. Am. Coll. Cardiol. 2004, 43, 1853–1860. [Google Scholar] [CrossRef] [Green Version]
- Mizusawa, Y.; Sakurada, H.; Nishizaki, M.; Hiraoka, M. Effects of low-dose quinidine on ventricular tachyarrhythmias in patients with Brugada syndrome: Low-dose quinidine therapy as an adjunctive treatment. J. Cardiovasc. Pharmacol. 2006, 47, 359–364. [Google Scholar] [CrossRef]
- Márquez, M.F.; Bonny, A.; Hernández-Castillo, E.; De Sisti, A.; Gómez-Flores, J.; Nava, S.; Hidden-Lucet, F.; Iturralde, P.; Cárdenas, M.; Tonet, J. Long-term efficacy of low doses of quinidine on malignant arrhythmias in Brugada syndrome with an implantable cardioverter-defibrillator: A case series and literature review. Heart Rhythm 2012, 9, 1995–2000. [Google Scholar] [CrossRef] [PubMed]
- Belhassen, B.; Glick, A.; Viskin, S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation 2004, 110, 1731–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, M.; Laidlaw, J.C.; Oshrain, C.; Willis, P.W.; Nissen, C.H.; McDermott, D.J.; Smith, W.S.; Karim, A.; Wilson, R.R. A randomized, double-blind, parallel group comparison of disopyramide phosphate and quinidine in patients with cardiac arrhythmias. Angiology 1983, 34, 393–400. [Google Scholar] [CrossRef]
- Naccarella, F.; Bracchetti, D.; Palmieri, M.; Cantelli, I.; Bertaccini, P.; Ambrosioni, E. Comparison of propafenone and disopyramide for treatment of chronic ventricular arrhythmias: Placebo-controlled, double-blind, randomized crossover study. Am. Heart J. 1985, 109, 833–840. [Google Scholar] [CrossRef]
- Sbarbaro, J.A.; Rawling, D.A.; Fozzard, H.A. Suppression of ventricular arrhythmias with intravenous disopyramide and lidocaine: Efficacy comparison in a randomized trial. Am. J. Cardiol. 1979, 44, 513–520. [Google Scholar] [CrossRef]
- Tanabe, T.; Takahashi, K.; Yoshioka, K.; Goto, Y. Evaluation of disopyramide and mexiletine used alone and in combination for ventricular arrhythmias in patients with and without overt heart disease. Int. J. Cardiol. 1991, 32, 303–312. [Google Scholar] [CrossRef]
- Ekelund, L.G.; Nilsson, E.; Walldius, G. Efficacy of and adverse effects of disopyramide. Comparison of capsules, controlled release tablets and placebo in patients with chronic ventricular arrhythmias. Eur. J. Clin. Pharmacol. 1986, 29, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Sherrid, M.V.; Shetty, A.; Winson, G.; Kim, B.; Musat, D.; Alviar, C.L.; Homel, P.; Balaram, S.K.; Swistel, D.G. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with β-blockade or verapamil. Circ. Heart Fail 2013, 6, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Adler, A.; Fourey, D.; Weissler-Snir, A.; Hindieh, W.; Chan, R.H.; Gollob, M.H.; Rakowski, H. Safety of Outpatient Initiation of Disopyramide for Obstructive Hypertrophic Cardiomyopathy Patients. J. Am. Heart Assoc. 2017, 6, e005152. [Google Scholar] [CrossRef]
- Rosen, M.R.; Hoffman, B.F.; Wit, A.L. Electrophysiology and pharmacology of cardiac arrhythmias. V. Cardiac antiarrhythmic effects of lidocaine. Am. Heart J. 1975, 89, 526–536. [Google Scholar] [CrossRef]
- Hine, L.K.; Laird, N.; Hewitt, P.; Chalmers, T.C. Meta-analytic evidence against prophylactic use of lidocaine in acute myocardial infarction. Arch. Intern. Med. 1989, 149, 2694–2698. [Google Scholar] [CrossRef]
- Glover, B.M.; Brown, S.P.; Morrison, L.; Davis, D.; Kudenchuk, P.J.; Van Ottingham, L.; Vaillancourt, C.; Cheskes, S.; Atkins, D.L.; Dorian, P.; et al. Wide variability in drug use in out-of-hospital cardiac arrest: A report from the resuscitation outcomes consortium. Resuscitation 2012, 83, 1324–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorian, P.; Cass, D.; Schwartz, B.; Cooper, R.; Gelaznikas, R.; Barr, A. Amiodarone as compared with lidocaine for shock-resistant ventricular fibrillation. N. Engl. J. Med. 2002, 346, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Kudenchuk, P.J.; Brown, S.P.; Daya, M.; Rea, T.; Nichol, G.; Morrison, L.J.; Leroux, B.; Vaillancourt, C.; Wittwer, L.; Callaway, C.W.; et al. Amiodarone, Lidocaine, or Placebo in Out-of-Hospital Cardiac Arrest. N. Engl. J. Med. 2016, 374, 1711–1722. [Google Scholar] [CrossRef]
- DiMarco, J.P.; Garan, H.; Ruskin, J.N. Mexiletine for refractory-ventricular arrhythmias: Results using serial electrophysiologic testing. Am. J. Cardiol. 1981, 47, 131–138. [Google Scholar] [CrossRef]
- Flaker, G.C.; Madigan, N.P.; Alpert, M.A.; Moser, S.A. Mexiletine for recurring ventricular arrhythmias: Assessment by long-term electrocardiographic recordings and sequential electrophysiologic studies. Am. Heart J. 1984, 108, 490–495. [Google Scholar] [CrossRef]
- Podrid, P.J.; Lown, B. Mexiletine for ventricular arrhythmias. Am. J. Cardiol. 1981, 47, 895–902. [Google Scholar] [CrossRef]
- Singh, S.; Klein, R.; Eisenberg, B.; Hughes, E.; Shand, M.; Doherty, P. Long-term effect of mexiletine on left ventricular function and relation to suppression of ventricular arrhythmia. Am. J. Cardiol. 1990, 66, 1222–1227. [Google Scholar] [CrossRef]
- Stein, J.; Podrid, P.J.; Lampert, S.; Hirsowitz, G.; Lown, B. Long-term mexiletine for ventricular arrhythmia. Am. Heart J. 1984, 107, 1091–1098. [Google Scholar] [CrossRef]
- Duke, M. Chronic mexiletine therapy for suppression of ventricular arrhythmias. Clin. Cardiol. 1988, 11, 132–136. [Google Scholar] [CrossRef]
- Claro, J.C.; Candia, R.; Rada, G.; Baraona, F.; Larrondo, F.; Letelier, L.M. Amiodarone versus other pharmacological interventions for prevention of sudden cardiac death. Cochrane Database Syst. Rev. 2015, CD008093. [Google Scholar] [CrossRef]
- Sobiech, M.; Lewandowski, M.; Zając, D.; Maciąg, A.; Syska, P.; Ateńska-Pawłowska, J.; Kowalik, I.; Sterliński, M.; Szwed, H.; Pytkowski, M. Efficacy and tolerability of mexiletine treatment in patients with recurrent ventricular tachyarrhythmias and implantable cardioverter-defibrillator shocks. Kardiol. Pol. 2017, 75, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Mugnai, G.; Paolini, C.; Cavedon, S.; Mecenero, A.; Perrone, C.; Bilato, C. Mexiletine for ventricular arrhythmias in patients with chronic coronary syndrome: A cohort study. Acta Cardiol. 2021, 77, 264–270. [Google Scholar] [CrossRef]
- Sapp, J.L.; Wells, G.A.; Parkash, R.; Stevenson, W.G.; Blier, L.; Sarrazin, J.F.; Thibault, B.; Rivard, L.; Gula, L.; Leong-Sit, P.; et al. Ventricular Tachycardia Ablation versus Escalation of Antiarrhythmic Drugs. N. Engl. J. Med. 2016, 375, 111–121. [Google Scholar] [CrossRef]
- Gao, D.; Van Herendael, H.; Alshengeiti, L.; Dorian, P.; Mangat, I.; Korley, V.; Ahmad, K.; Golovchiner, G.; Aves, T.; Pinter, A. Mexiletine as an adjunctive therapy to amiodarone reduces the frequency of ventricular tachyarrhythmia events in patients with an implantable defibrillator. J. Cardiovasc. Pharmacol. 2013, 62, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Maragna, R.; Faragli, A.; Monteforte, N.; Bloise, R.; Memmi, M.; Novelli, V.; Baiardi, P.; Bagnardi, V.; Etheridge, S.P.; et al. Gene-Specific Therapy With Mexiletine Reduces Arrhythmic Events in Patients With Long QT Syndrome Type 3. J. Am. Coll. Cardiol. 2016, 67, 1053–1058. [Google Scholar] [CrossRef]
- Holmes, B.; Heel, R.C. Flecainide. Drugs 1985, 29, 1–33. [Google Scholar] [CrossRef]
- Kuck, K.H.; Cappato, R.; Siebels, J.; Rüppel, R. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest: The Cardiac Arrest Study Hamburg (CASH). Circulation 2000, 102, 748–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echt, D.S.; Liebson, P.R.; Mitchell, L.B.; Peters, R.W.; Obias-Manno, D.; Barker, A.H.; Arensberg, D.; Baker, A.; Friedman, L.; Greene, H.L. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med. 1991, 324, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Roston, T.M.; Vinocur, J.M.; Maginot, K.R.; Mohammed, S.; Salerno, J.C.; Etheridge, S.P.; Cohen, M.; Hamilton, R.M.; Pflaumer, A.; Kanter, R.J.; et al. Catecholaminergic polymorphic ventricular tachycardia in children: Analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ. Arrhythm. Electrophysiol. 2015, 8, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Khoury, A.; Marai, I.; Suleiman, M.; Blich, M.; Lorber, A.; Gepstein, L.; Boulos, M. Flecainide therapy suppresses exercise-induced ventricular arrhythmias in patients with CASQ2-associated catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2013, 10, 1671–1675. [Google Scholar] [CrossRef]
- Belardinelli, L.; Giles, W.R.; Rajamani, S.; Karagueuzian, H.S.; Shryock, J.C. Cardiac late Na⁺ current: Proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm 2015, 12, 440–448. [Google Scholar] [CrossRef]
- Moss, A.J.; Windle, J.R.; Hall, W.J.; Zareba, W.; Robinson, J.L.; McNitt, S.; Severski, P.; Rosero, S.; Daubert, J.P.; Qi, M.; et al. Safety and efficacy of flecainide in subjects with Long QT-3 syndrome (DeltaKPQ mutation): A randomized, double-blind, placebo-controlled clinical trial. Ann. Noninvasive Electrocardiol. 2005, 10, 59–66. [Google Scholar] [CrossRef]
- Grandi, E.; Ripplinger, C.M. Antiarrhythmic mechanisms of beta blocker therapy. Pharmacol. Res. 2019, 146, 104274. [Google Scholar] [CrossRef]
- Al-Gobari, M.; El Khatib, C.; Pillon, F.; Gueyffier, F. β-Blockers for the prevention of sudden cardiac death in heart failure patients: A meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2013, 13, 52. [Google Scholar] [CrossRef] [Green Version]
- Exner, D.V.; Reiffel, J.A.; Epstein, A.E.; Ledingham, R.; Reiter, M.J.; Yao, Q.; Duff, H.J.; Follmann, D.; Schron, E.; Greene, H.L.; et al. Beta-blocker use and survival in patients with ventricular fibrillation or symptomatic ventricular tachycardia: The Antiarrhythmics Versus Implantable Defibrillators (AVID) trial. J. Am. Coll. Cardiol. 1999, 34, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Exner, D.V.; Dries, D.L.; Waclawiw, M.A.; Shelton, B.; Domanski, M.J. Beta-adrenergic blocking agent use and mortality in patients with asymptomatic and symptomatic left ventricular systolic dysfunction: A post hoc analysis of the Studies of Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 1999, 33, 916–923. [Google Scholar] [CrossRef] [Green Version]
- McMurray, J.; Køber, L.; Robertson, M.; Dargie, H.; Colucci, W.; Lopez-Sendon, J.; Remme, W.; Sharpe, D.N.; Ford, I. Antiarrhythmic effect of carvedilol after acute myocardial infarction: Results of the Carvedilol Post-Infarct Survival Control in Left Ventricular Dysfunction (CAPRICORN) trial. J. Am. Coll. Cardiol. 2005, 45, 525–530. [Google Scholar] [CrossRef] [Green Version]
- BHAT Study Investigators. A randomized trial of propranolol in patients with acute myocardial infarction. I. Mortality results. JAMA 1982, 247, 1707–1714. [Google Scholar] [CrossRef]
- Woosley, R.L.; Kornhauser, D.; Smith, R.; Reele, S.; Higgins, S.B.; Nies, A.S.; Shand, D.G.; Oates, J.A. Suppression of chronic ventricular arrhythmias with propranolol. Circulation 1979, 60, 819–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodsky, M.A.; Allen, B.J.; Luckett, C.R.; Capparelli, E.V.; Wolff, L.J.; Henry, W.L. Antiarrhythmic efficacy of solitary beta-adrenergic blockade for patients with sustained ventricular tachyarrhythmias. Am. Heart J. 1989, 118, 272–280. [Google Scholar] [CrossRef]
- Levine, J.H.; Mellits, E.D.; Baumgardner, R.A.; Veltri, E.P.; Mower, M.; Grunwald, L.; Guarnieri, T.; Aarons, D.; Griffith, L.S. Predictors of first discharge and subsequent survival in patients with automatic implantable cardioverter-defibrillators. Circulation 1991, 84, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Connolly, S.J.; Dorian, P.; Roberts, R.S.; Gent, M.; Bailin, S.; Fain, E.S.; Thorpe, K.; Champagne, J.; Talajic, M.; Coutu, B.; et al. Comparison of β-Blockers, Amiodarone Plus β-Blockers, or Sotalol for Prevention of Shocks From Implantable Cardioverter DefibrillatorsThe OPTIC Study: A Randomized Trial. JAMA 2006, 295, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Boutitie, F.; Boissel, J.P.; Connolly, S.J.; Camm, A.J.; Cairns, J.A.; Julian, D.G.; Gent, M.; Janse, M.J.; Dorian, P.; Frangin, G. Amiodarone interaction with beta-blockers: Analysis of the merged EMIAT (European Myocardial Infarct Amiodarone Trial) and CAMIAT (Canadian Amiodarone Myocardial Infarction Trial) databases. The EMIAT and CAMIAT Investigators. Circulation 1999, 99, 2268–2275. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, H.L.; Brooks, M.M.; Barker, A.H.; Bergstrand, R.; Huther, M.L.; Beanlands, D.S.; Bigger, J.T.; Goldstein, S. Beta-blocker therapy in the Cardiac Arrhythmia Suppression Trial. CAST Investigators. Am. J. Cardiol. 1994, 74, 674–680. [Google Scholar] [CrossRef]
- Hirsowitz, G.; Podrid, P.J.; Lampert, S.; Stein, J.; Lown, B. The role of beta blocking agents as adjunct therapy to membrane stabilizing drugs in malignant ventricular arrhythmia. Am. Heart J. 1986, 111, 852–860. [Google Scholar] [CrossRef]
- Szabó, B.M.; Crijns, H.J.; Wiesfeld, A.C.; van Veldhuisen, D.J.; Hillege, H.L.; Lie, K.I. Predictors of mortality in patients with sustained ventricular tachycardias or ventricular fibrillation and depressed left ventricular function: Importance of beta-blockade. Am. Heart J. 1995, 130, 281–286. [Google Scholar] [CrossRef]
- Reiter, M.J.; Reiffel, J.A. Importance of beta blockade in the therapy of serious ventricular arrhythmias. Am. J. Cardiol. 1998, 82, 9I–19I. [Google Scholar] [CrossRef]
- Muser, D.; Santangeli, P.; Liang, J.J. Management of ventricular tachycardia storm in patients with structural heart disease. World J. Cardiol. 2017, 9, 521–530. [Google Scholar] [CrossRef]
- Nademanee, K.; Taylor, R.; Bailey, W.E.; Rieders, D.E.; Kosar, E.M. Treating electrical storm: Sympathetic blockade versus advanced cardiac life support-guided therapy. Circulation 2000, 102, 742–747. [Google Scholar] [CrossRef] [Green Version]
- Chatzidou, S.; Kontogiannis, C.; Tsilimigras, D.I.; Georgiopoulos, G.; Kosmopoulos, M.; Papadopoulou, E.; Vasilopoulos, G.; Rokas, S. Propranolol Versus Metoprolol for Treatment of Electrical Storm in Patients With Implantable Cardioverter-Defibrillator. J. Am. Coll. Cardiol. 2018, 71, 1897–1906. [Google Scholar] [CrossRef]
- Puljević, M.; Velagić, V.; Puljević, D.; Miličić, D. Propranolol efficiency in prevention of sustained ventricular tachycardia in patients with implanted cardioverter-defibrillator: A case series. Croat. Med. J. 2014, 55, 75–76. [Google Scholar] [CrossRef] [Green Version]
- Huikuri, H.V.; Cox, M.; Interian, A.; Kessler, K.M.; Glicksman, F.; Castellanos, A.; Myerburg, R.J. Efficacy of intravenous propranolol for suppression of inducibility of ventricular tachyarrhythmias with different electrophysiologic characteristics in coronary artery disease. Am. J. Cardiol. 1989, 64, 1305–1309. [Google Scholar] [CrossRef]
- Hayashi, M.; Denjoy, I.; Extramiana, F.; Maltret, A.; Buisson, N.R.; Lupoglazoff, J.M.; Klug, D.; Takatsuki, S.; Villain, E.; Kamblock, J.; et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation 2009, 119, 2426–2434. [Google Scholar] [CrossRef] [Green Version]
- Leenhardt, A.; Lucet, V.; Denjoy, I.; Grau, F.; Ngoc, D.D.; Coumel, P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 1995, 91, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013, 15, 1389–1406. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Locati, E. The idiopathic long QT syndrome: Pathogenetic mechanisms and therapy. Eur. Heart J. 1985, 6 (Suppl. D), 103–114. [Google Scholar] [CrossRef]
- Bennett, M.T.; Gula, L.J.; Klein, G.J.; Skanes, A.C.; Yee, R.; Leong-Sit, P.; Chattha, I.; Sy, R.; Jones, D.L.; Krahn, A.D. Effect of beta-blockers on QT dynamics in the long QT syndrome: Measuring the benefit. Europace 2014, 16, 1847–1851. [Google Scholar] [CrossRef]
- Wilde, A.A.; Moss, A.J.; Kaufman, E.S.; Shimizu, W.; Peterson, D.R.; Benhorin, J.; Lopes, C.; Towbin, J.A.; Spazzolini, C.; Crotti, L.; et al. Clinical Aspects of Type 3 Long-QT Syndrome: An International Multicenter Study. Circulation 2016, 134, 872–882. [Google Scholar] [CrossRef] [Green Version]
- Woosley, R.L.; Funck-Brentano, C. Overview of the clinical pharmacology of antiarrhythmic drugs. Am. J. Cardiol. 1988, 61, A61–A69. [Google Scholar] [CrossRef]
- Mattioni, T.A.; Zheutlin, T.A.; Sarmiento, J.J.; Parker, M.; Lesch, M.; Kehoe, R.F. Amiodarone in patients with previous drug-mediated torsade de pointes. Long-term safety and efficacy. Ann. Intern. Med. 1989, 111, 574–580. [Google Scholar] [CrossRef]
- Torres, V.; Tepper, D.; Flowers, D.; Wynn, J.; Lam, S.; Keefe, D.; Miura, D.S.; Somberg, J.C. QT prolongation and the antiarrhythmic efficacy of amiodarone. J. Am. Coll. Cardiol. 1986, 7, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Julian, D.G.; Camm, A.J.; Frangin, G.; Janse, M.J.; Munoz, A.; Schwartz, P.J.; Simon, P. Randomised trial of effect of amiodarone on mortality in patients with left-ventricular dysfunction after recent myocardial infarction: EMIAT. European Myocardial Infarct Amiodarone Trial Investigators. Lancet 1997, 349, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Cairns, J.A.; Connolly, S.J.; Roberts, R.; Gent, M. Randomised trial of outcome after myocardial infarction in patients with frequent or repetitive ventricular premature depolarisations: CAMIAT. Canadian Amiodarone Myocardial Infarction Arrhythmia Trial Investigators. Lancet 1997, 349, 675–682. [Google Scholar] [CrossRef]
- Greene, H.L. The CASCADE Study: Randomized antiarrhythmic drug therapy in survivors of cardiac arrest in Seattle. CASCADE Investigators. Am. J. Cardiol. 1993, 72, 70F–74F. [Google Scholar] [CrossRef]
- Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: Meta-analysis of individual data from 6500 patients in randomised trials. Amiodarone Trials Meta-Analysis Investigators. Lancet 1997, 350, 1417–1424. [Google Scholar] [CrossRef]
- Kowey, P.R.; Crijns, H.J.; Aliot, E.M.; Capucci, A.; Kulakowski, P.; Radzik, D.; Roy, D.; Connolly, S.J.; Hohnloser, S.H.; Investigators, A.S. Efficacy and safety of celivarone, with amiodarone as calibrator, in patients with an implantable cardioverter-defibrillator for prevention of implantable cardioverter-defibrillator interventions or death: The ALPHEE study. Circulation 2011, 124, 2649–2660. [Google Scholar] [CrossRef]
- Connolly, S.J.; Hallstrom, A.P.; Cappato, R.; Schron, E.B.; Kuck, K.H.; Zipes, D.P.; Greene, H.L.; Boczor, S.; Domanski, M.; Follmann, D.; et al. Meta-analysis of the implantable cardioverter defibrillator secondary prevention trials. AVID, CASH and CIDS studies. Antiarrhythmics vs Implantable Defibrillator study. Cardiac Arrest Study Hamburg. Canadian Implantable Defibrillator Study. Eur. Heart J. 2000, 21, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M.; Keam, S.J. Dronedarone. Drugs 2009, 69, 1647–1663. [Google Scholar] [CrossRef]
- Tave, A.; Goehring, E.; Desai, V.; Wu, C.; Bohn, R.L.; Tamayo, S.G.; Sicignano, N.; Juhaeri, J.; Jones, J.K.; Weiss, S.R. Risk of interstitial lung disease in patients treated for atrial fibrillation with dronedarone versus other antiarrhythmics. Pharmacoepidemiol. Drug Saf. 2021, 30, 1353–1359. [Google Scholar] [CrossRef]
- Friberg, L. Ventricular arrhythmia and death among atrial fibrillation patients using anti-arrhythmic drugs. Am. Heart J. 2018, 205, 118–127. [Google Scholar] [CrossRef]
- Køber, L.; Torp-Pedersen, C.; McMurray, J.J.; Gøtzsche, O.; Lévy, S.; Crijns, H.; Amlie, J.; Carlsen, J.; Group, D.S. Increased mortality after dronedarone therapy for severe heart failure. N. Engl. J. Med. 2008, 358, 2678–2687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalus, J.S.; Mauro, V.F. Dofetilide: A Class III-Specific Antiarrhythmic Agent. Ann. Pharmacother. 2000, 34, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Mounsey, J.P.; DiMarco, J.P. Dofetilide. Circulation 2000, 102, 2665–2670. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Zoble, R.G.; Yellen, L.; Brodsky, M.A.; Feld, G.K.; Berk, M.; Billing, C.B. Efficacy and Safety of Oral Dofetilide in Converting to and Maintaining Sinus Rhythm in Patients With Chronic Atrial Fibrillation or Atrial Flutter. Circulation 2000, 102, 2385–2390. [Google Scholar] [CrossRef] [Green Version]
- Baquero, G.A.; Banchs, J.E.; Depalma, S.; Young, S.K.; Penny-Peterson, E.D.; Samii, S.M.; Wolbrette, D.L.; Naccarelli, G.V.; Gonzalez, M.D. Dofetilide Reduces the Frequency of Ventricular Arrhythmias and Implantable Cardioverter Defibrillator Therapies. J. Cardiovasc. Electrophysiol. 2012, 23, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Torp-Pedersen, C.; Møller, M.; Bloch-Thomsen, P.E.; Køber, L.; Sandøe, E.; Egstrup, K.; Agner, E.; Carlsen, J.; Videbæk, J.; Marchant, B.; et al. Dofetilide in Patients with Congestive Heart Failure and Left Ventricular Dysfunction. N. Engl. J. Med. 1999, 341, 857–865. [Google Scholar] [CrossRef]
- Køber, L.; Thomsen, P.E.B.; Møller, M.; Torp-Pedersen, C.; Carlsen, J.; Sandøe, E.; Egstrup, K.; Agner, E.; Videbæk, J.; Marchant, B.; et al. Effect of dofetilide in patients with recent myocardial infarction and left-ventricular dysfunction: A randomised trial. Lancet 2000, 356, 2052–2058. [Google Scholar] [CrossRef]
- Echt, D.S.; Lee, J.T.; Murray, K.T.; Vorperian, V.; Borganeeli, S.M.; Crawford, D.M.; Friedrich, T.; Roden, D.M. A Randomized, Double-Blind, Placebo-Controlled, Dose-Ranging Study of Dofetilide in Patients with Inducible Sustained Ventricular Tachyarrhythmias. J. Cardiovasc. Electrophysiol. 1995, 6, 687–699. [Google Scholar] [CrossRef]
- Bashir, Y.; Thomsen, P.-E.B.; Kingma, J.H.; Møller, M.; Wong, C.; Cobbe, S.M.; Jordaens, L.; Campbell, R.W.F.; Rasmussen, H.S.; Camm, A.J. Electrophysiologic profile and efficacy of intravenous dofetilide (UK-68,798), a new class III antiarrhythmic drug, in patients with sustained monomorphic ventricular tachycardia. Am. J. Cardiol. 1995, 76, 1040–1044. [Google Scholar] [CrossRef]
- Boriani, G.; Lubinski, A.; Capucci, A.; Niederle, P.; Kornacewicz-Jack, Z.; Wnuk-Wojnar, A.M.; Borggrefe, M.; Brachmann, J.; Biffi, M.; Butrous, G.S.; et al. A multicentre, double-blind randomized crossover comparative study on the efficacy and safety of dofetilide vs sotalol in patients with inducible sustained ventricular tachycardia and ischaemic heart disease. Eur. Heart J. 2001, 22, 2180–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.L.; Prystowsky, E.N. Sotalol: An important new antiarrhythmic. Am. Heart J. 1999, 137, 388–409. [Google Scholar] [CrossRef]
- Anastasiou-Nana, M.I.; Gilbert, E.M.; Miller, R.H.; Singh, S.; Freedman, R.A.; Keefe, D.L.; Saksena, S.; MacNeil, D.J.; Anderson, J.L. Usefulness of d, I sotalol for suppression of chronic ventricular arrhythmias. Am. J. Cardiol. 1991, 67, 511–516. [Google Scholar] [CrossRef]
- Roden, D.M. Usefulness of sotalol for life-threatening ventricular arrhythmias. Am. J. Cardiol. 1993, 72, A51–A55. [Google Scholar] [CrossRef]
- Hohnloser, S.H.; Meinertz, T.; Stubbs, P.; Crijns, H.J.G.M.; Blanc, J.-J.; Rizzon, P.; Cheuvart, B. Efficacy and Safety of d-Sotalol, a Pure Class III Antiarrhythmic Compound, in Patients With Symptomatic Complex Ventricular Ectopy. Circulation 1995, 92, 1517–1525. [Google Scholar] [CrossRef]
- Waldo, A.L.; Camm, A.J.; deRuyter, H.; Friedman, P.L.; MacNeil, D.J.; Pauls, J.F.; Pitt, B.; Pratt, C.M.; Schwartz, P.J.; Veltri, E.P. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. Lancet 1996, 348, 7–12. [Google Scholar] [CrossRef]
- Pacifico, A.; Hohnloser, S.H.; Williams, J.H.; Tao, B.; Saksena, S.; Henry, P.D.; Prystowsky, E.N. Prevention of Implantable-Defibrillator Shocks by Treatment with Sotalol. N. Engl. J. Med. 1999, 340, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Wichter, T.; Borggrefe, M.; Haverkamp, W.; Chen, X.; Breithardt, G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation 1992, 86, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.N.; Nademanee, K. Use of calcium antagonists for cardiac arrhythmias. Am. J. Cardiol. 1987, 59, B153–B162. [Google Scholar] [CrossRef]
- Wellens, H.J.J.; Bär, F.W.H.M.; Lie, K.I.; Düren, D.R.; Dohmen, H.J. Effect of procainamide, propranolol and verapamil on mechanism of tachycardia in patients with chronic recurrent ventricular tachycardia. Am. J. Cardiol. 1977, 40, 579–585. [Google Scholar] [CrossRef]
- Belhassen, B.; Horowitz, L.N. Use of intravenous verapamil for ventricular tachycardia. Am. J. Cardiol. 1984, 54, 1131–1133. [Google Scholar] [CrossRef]
- Belhassen, B.; Rotmensch, H.H.; Laniado, S. Response of recurrent sustained ventricular tachycardia to verapamil. Br. Heart J. 1981, 46, 679–682. [Google Scholar] [CrossRef]
- Ohe, T.; Shimomura, K.; Aihara, N.; Kamakura, S.; Matsuhisa, M.; Sato, I.; Nakagawa, H.; Shimizu, A. Idiopathic sustained left ventricular tachycardia: Clinical and electrophysiologic characteristics. Circulation 1988, 77, 560–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogami, A.; Naito, S.; Tada, H.; Taniguchi, K.; Okamoto, Y.; Nishimura, S.; Yamauchi, Y.; Aonuma, K.; Goya, M.; Iesaka, Y.; et al. Demonstration of diastolic and presystolic purkinje potentials as critical potentials in a macroreentry circuit of verapamil-sensitive idiopathic left ventricular tachycardia. J. Am. Coll. Cardiol. 2000, 36, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Van der Werf, C.; Zwinderman, A.H.; Wilde, A.A.M. Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: State of the art and future developments. EP Eur. 2011, 14, 175–183. [Google Scholar] [CrossRef]
- Rosso, R.; Kalman, J.M.; Rogowski, O.; Diamant, S.; Birger, A.; Biner, S.; Belhassen, B.; Viskin, S. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2007, 4, 1149–1154. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Belardinelli, L.; Zygmunt, A.C.; Burashnikov, A.; Diego, J.M.D.; Fish, J.M.; Cordeiro, J.M.; Thomas, G. Electrophysiological Effects of Ranolazine, a Novel Antianginal Agent With Antiarrhythmic Properties. Circulation 2004, 110, 904–910. [Google Scholar] [CrossRef]
- Rayner-Hartley, E.; Sedlak, T. Ranolazine: A Contemporary Review. J. Am. Heart Assoc 2016, 5, e003196. [Google Scholar] [CrossRef] [Green Version]
- Zareba, W.; Daubert, J.P.; Beck, C.A.; Huang, D.T.; Alexis, J.D.; Brown, M.W.; Pyykkonen, K.; McNitt, S.; Oakes, D.; Feng, C.; et al. Ranolazine in High-Risk Patients With Implanted Cardioverter-Defibrillators: The RAID Trial. J. Am. Coll. Cardiol. 2018, 72, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.A.; Scirica, B.M.; Karwatowska-Prokopczuk, E.; Murphy, S.A.; Budaj, A.; Varshavsky, S.; Wolff, A.A.; Skene, A.; McCabe, C.H.; Braunwald, E.; et al. Effects of ranolazine on recurrent cardiovascular events in patients with non-ST-elevation acute coronary syndromes: The MERLIN-TIMI 36 randomized trial. JAMA 2007, 297, 1775–1783. [Google Scholar] [CrossRef] [Green Version]
- Bunch, T.J.; Mahapatra, S.; Murdock, D.; Molden, J.; Weiss, J.P.; May, H.T.; Bair, T.L.; Mader, K.M.; Crandall, B.G.; Day, J.D.; et al. Ranolazine reduces ventricular tachycardia burden and ICD shocks in patients with drug-refractory ICD shocks. Pacing Clin. Electrophysiol. 2011, 34, 1600–1606. [Google Scholar] [CrossRef]
- Olivotto, I.; Camici, P.G.; Merlini, P.A.; Rapezzi, C.; Patten, M.; Climent, V.; Sinagra, G.; Tomberli, B.; Marin, F.; Ehlermann, P.; et al. Efficacy of Ranolazine in Patients with Symptomatic Hypertrophic Cardiomyopathy: The RESTYLE-HCM Randomized, Double-Blind, Placebo-Controlled Study. Circ. Heart Fail 2018, 11, e004124. [Google Scholar] [CrossRef]
- Lerman, B.B.; Stein, K.M.; Markowitz, S.M.; Mittal, S.; Slotwiner, D.J. Right ventricular outflow tract tachycardia: An update. Card. Electrophysiol. Rev. 2002, 6, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.M.; Litvak, B.L.; Ramirez de Arellano, E.A.; Markisz, J.A.; Stein, K.M.; Lerman, B.B. Adenosine-sensitive ventricular tachycardia: Right ventricular abnormalities delineated by magnetic resonance imaging. Circulation 1997, 96, 1192–1200. [Google Scholar] [CrossRef]
- Akdis, D.; Brunckhorst, C.; Duru, F.; Saguner, A.M. Arrhythmogenic Cardiomyopathy: Electrical and Structural Phenotypes. Arrhythm Electrophysiol. Rev. 2016, 5, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerman, B.B.; Ip, J.E.; Shah, B.K.; Thomas, G.; Liu, C.F.; Ciaccio, E.J.; Wit, A.L.; Cheung, J.W.; Markowitz, S.M. Mechanism-specific effects of adenosine on ventricular tachycardia. J. Cardiovasc. Electrophysiol. 2014, 25, 1350–1358. [Google Scholar] [CrossRef]
- Group, D.I. The effect of digoxin on mortality and morbidity in patients with heart failure. N. Engl. J. Med. 1997, 336, 525–533. [Google Scholar] [CrossRef]
- Ziff, O.J.; Lane, D.A.; Samra, M.; Griffith, M.; Kirchhof, P.; Lip, G.Y.; Steeds, R.P.; Townend, J.; Kotecha, D. Safety and efficacy of digoxin: Systematic review and meta-analysis of observational and controlled trial data. BMJ 2015, 351, h4451. [Google Scholar] [CrossRef] [Green Version]
- Vamos, M.; Erath, J.W.; Hohnloser, S.H. Digoxin-associated mortality: A systematic review and meta-analysis of the literature. Eur. Heart J. 2015, 36, 1831–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.S.; Hardy, J.; Yee, R.; Healey, J.S.; Birnie, D.; Simpson, C.S.; Crystal, E.; Mangat, I.; Nanthakumar, K.; Wang, X.; et al. Clinical Risk Stratification for Primary Prevention Implantable Cardioverter Defibrillators. Circ. Heart Fail. 2015, 8, 927–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.Y.; Kutyifa, V.; Ruwald, M.H.; McNitt, S.; Polonsky, B.; Zareba, W.; Moss, A.J.; Ruwald, A.C. Digoxin therapy and associated clinical outcomes in the MADIT-CRT trial. Heart Rhythm 2015, 12, 2010–2017. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kinoshita, O.; Uchikawa, S.; Kasai, H.; Nakamura, M.; Izawa, A.; Yokoseki, O.; Kitabayashi, H.; Takahashi, W.; Yazaki, Y.; et al. Successful prevention of recurrent ventricular fibrillation by intravenous isoproterenol in a patient with Brugada syndrome. Pacing Clin. Electrophysiol. 2001, 24, 1293–1294. [Google Scholar] [CrossRef]
- Ohgo, T.; Okamura, H.; Noda, T.; Satomi, K.; Suyama, K.; Kurita, T.; Aihara, N.; Kamakura, S.; Ohe, T.; Shimizu, W. Acute and chronic management in patients with Brugada syndrome associated with electrical storm of ventricular fibrillation. Heart Rhythm 2007, 4, 695–700. [Google Scholar] [CrossRef]
- Koruth, J.S.; Lala, A.; Pinney, S.; Reddy, V.Y.; Dukkipati, S.R. The Clinical Use of Ivabradine. J. Am. Coll. Cardiol. 2017, 70, 1777–1784. [Google Scholar] [CrossRef]
- Fox, K.; Ford, I.; Steg, P.G.; Tardif, J.-C.; Tendera, M.; Ferrari, R. Ivabradine in Stable Coronary Artery Disease without Clinical Heart Failure. N. Engl. J. Med. 2014, 371, 1091–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, K.; Ford, I.; Steg, P.G.; Tendera, M.; Ferrari, R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 807–816. [Google Scholar] [CrossRef]
- Swedberg, K.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef]
- Cappato, R.; Castelvecchio, S.; Ricci, C.; Bianco, E.; Vitali-Serdoz, L.; Gnecchi-Ruscone, T.; Pittalis, M.; De Ambroggi, L.; Baruscotti, M.; Gaeta, M.; et al. Clinical Efficacy of Ivabradine in Patients With Inappropriate Sinus Tachycardia: A Prospective, Randomized, Placebo-Controlled, Double-Blind, Crossover Evaluation. J. Am. Coll. Cardiol. 2012, 60, 1323–1329. [Google Scholar] [CrossRef] [Green Version]
- Kohli, U.; Aziz, Z.; Beaser, A.D.; Nayak, H.M. Ventricular arrhythmia suppression with ivabradine in a patient with catecholaminergic polymorphic ventricular tachycardia refractory to nadolol, flecainide, and sympathectomy. Pacing Clin. Electrophysiol. 2020, 43, 527–533. [Google Scholar] [CrossRef]
- Vaksmann, G.; Klug, D. Efficacy of ivabradine to control ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Pacing Clin. Electrophysiol. 2018, 41, 1378–1380. [Google Scholar] [CrossRef]
- Martin, R.I.R.; Pogoryelova, O.; Koref, M.S.; Bourke, J.P.; Teare, M.D.; Keavney, B.D. Atrial fibrillation associated with ivabradine treatment: Meta-analysis of randomised controlled trials. Heart 2014, 100, 1506–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocco, G.; Jerie, P. Torsades de Pointes Induced by the Concomitant Use of Ivabradine and Azithromycin: An Unexpected Dangerous Interaction. Cardiovasc. Toxicol. 2015, 15, 104–106. [Google Scholar] [CrossRef]


{kind=link}
| Drug | Mechanism of Action | Channels Effected | Dosing | Contraindications | Important Side Effects/Considerations |
|---|---|---|---|---|---|
| Class I—Sodium Channel Agents | |||||
| Class IA | |||||
| Quinidine | • Blockade of the rapid inward sodium depolarization current in a use dependent fashion and prolongation of repolarization via blockade of the delayed rectifier potassium channel in reverse use-dependence fashion. | INa, Ito, IKr, M, α | PO only: Sulfate 300 mg-max tolerated q6h–q12h, Gluconate 324–648 mg q8h–q12h | Severe AV node dysfunction, Thrombocytopenia or underlying platelet dysfunction. Prolonged QT. | • Negative Ionotropy, profound hypotension. |
| • They slow phase 4 depolarization during spontaneous automaticity. | • Can be pro-arrhythmogenic at therapeutic doses | ||||
| • The net effect is preferential prolongation of action potential duration at fast heart rates, prolonged effective refractory period (ERP), and decreased automaticity | • Can prolong QT | ||||
| Procainamide | INa, IKr | IV: 10–17 mg/kg; PO: 500–1000 mg q6h | Severe AV node dysfunction, underlying Systemic Lupus Erythematosus. Prolonged QT. | • Can cause Drug-induced Systemic Lupus Erythematosus. | |
| • Can cause agranulocytosis at therapeutic doses requrining CBC monitoring | |||||
| • Breaks down into toxic metabolite ‘NAPA’ which requires monitoring, especially when on IV formulations. | |||||
| • Can cause negative inotropy and profound hypotension. | |||||
| • Can be pro-arrhythmogenic at therapeutic doses. | |||||
| • Can prolong QT | |||||
| Disopyramide | INa, Ito, IKr, IK(ATP), M | PO only: 150 mg q6h | Severe AV node dysfunction | • Negative Ionotropy, profound hypotension. | |
| • Can be pro-arrhythmogenic at therapeutic doses | |||||
| • Significant Anticholinergic side effects | |||||
| • Can prolong QT | |||||
| Class IB | |||||
| Lidocaine | • Use dependent blockade of the inward sodium depolarization current thereby decreasing maximal velocity of depolarization. | INa | IV only: 1 mg/kg bolus followed by 1–3 mg/min | Severe AV node dysfunction | • CNS side effects including seizures, coma, or death requiring frequent blood level monitoring. |
| Mexiletine | • Shortening of action potential and ERP duration thereby decreasing automaticity of phase 4 depolarization. | INa | PO only: 150 mg q8h | • Higher incidence of Drug-Induced Liver Injury. | |
| • Can cause tremors and ataxia | |||||
| • Has high rates of GI distress. | |||||
| Class IC | |||||
| Flecainide | • Most potent among the inward sodium blocking agents thereby markedly reducing the action potential conduction velocity in atrial, ventricular, and specialized conduction tissues. | INa, IKr, IKur | PO only: 50–200 mg q12h (can increase to q8h) | Structural Heart Disease or Reduced Ejection Fraction | • Can be pro-arrhythmogenic at therapeutic doses. |
| • Blocking occurs in a use-dependent fashion with minimal effect on overall action potential duration or ERP. | • PR and QRS prolongation | ||||
| Propafenone | INa, IKr, IKur, β, α | PO only: IR release 150–300 mg q8h; ER release: 225–425 mg q12h | • May cause the slowing of atrial arrhythmias leading to dangerous 1:1 conduction. | ||
| Class II—Beta Blockers | |||||
| Propranolol | • Blunting sympathetic activity on cardiac tissue, most notably through decreasing phase 4 depolarization thereby decreasing automaticity via decreased conduction velocity and increased ERP within the AV-node decreasing reentry | β1, β2, INa | IV: 1–3 mg boluses q5min, PO: 10–160 mg q6h–q12h | Severe AV node dysfunction, Sick Sinus Syndrome | • Can cause severe bradycardia and precipitate cardiogenic shock |
| Metoprolol | β1 | IV: 5mg q5min ×3, PO: Tartrate 12.5–200 mg q6h–q12h, Succinate 12.5–200 mg q12h–q24h | |||
| Nadolol | β1, β2 | PO only: 40–320 mg qDay | |||
| Carvedilol | β1, β2, α | PO only: 3.125–25 mg q12h | |||
| Class III—Potassium Channel Agents | |||||
| Amiodarone | • Primarily through blockade of the delayed rectifier potassium channel effectively prolonging repolarization and increasing the ERP to decrease the likelihood of reentry. | INa, ICa, IKr, IK1, IKs, Ito, β, α | IV: 150–300 mg bolus, 0.5–1 mg/min (1 mg/min for 6 h then 0.5 mg/min for 18 h), PO: initial 400 mg q12h then taper to as low as 100 mg q24h if needed | Pre-existing Thyroid, Liver, and Pulmonary Disease | • Requires biannual TSH and LFT monitoring for thyroid/liver toxicity. |
| • Additionally, amiodarone has mechanistic overlap with all other classes of antiarrhythmics and has vasodilatory and negative inotropic effects. | • Requires annual PFT monitoring for pulmonary fibrosis. | ||||
| • Can cause skin photosensitivity. | |||||
| • Can cause corneal microdeposits effecting vision | |||||
| Sotalol | • Sotalol is a racemic mixture of d- and l- sotalol with unique pharmacologic effects exhibiting class II (nonselective ß-blocker) properties. | IKr, β1, β2 | IV: 75 mg q12h, PO: 80–120 mg q12h | Prolonged QT | • Profound QT prolongation, must complete load under observation in hospital with EKG monitoring (although emerging data to support rapid IV loading) |
| • Additionally, to class III inhibition of delayed potassium rectifier channel resulting in an increase in action potential duration and effective refractory period. | • In patients with advanced heart failure, Sotalol can precipitate cardiogenic shock. | ||||
| Dofetilide | • Specific class III antiarrhythmic which blocks the delayed outward rectifying potassium current thereby increasing the effective refractory period (ERP) in a reverse use-dependence fashion without delaying intracardiac conduction. | IKr | PO only: 500 mcg q12h | ||
| Class IV—Calcium Channel Blockers | |||||
| Verapamil | • The non-dihydropyridine calcium channel antagonists, verapamil and diltiazem, exhibit antiarrhythmic effects predominately at the AV-node via blocking of slow inward Ca current. | ICa-L | IV: 2.5–5 mg q15–30 mins as tolerated, PO: IR release 120 mg q8h; ER release: 120–480 mg q12h–q24h | Severely Depressed EF (<35%), Severe AV node dysfunction | • Negative inotropy, can precipitate cardiogenic shock. |
| • Blocking of inward Ca current thereby prolongs the effective refractory period (ERP) with minimal effects on atrial/ventricular myocytes or the his-purkinje system | |||||
| Diltiazem | • Although less common, these agents can cause blockade of slow inward calcium channels on some sensitive fascicular tissues. | ICa-L | IV: 0.25 mg/kg bolus followed by 5–15 mg/h as tolerated, PO: IR release: 30–120 mg q6h–q12h, ER release: 30–240 mg q12h–q24h | Severely Depressed EF (<35%), Severe AV node dysfunction | • Negative inotropy, can precipitate cardiogenic shock. |
| No Class in Vaughn-Williams | |||||
| Ranolazine | • Ranolazine exhibits features most similar to amiodarone, blocking inward depolarizing and outward repolarizing currents affecting sodium, potassium, and calcium channels. | INa, IKr | PO only: 500–1000 mg q12h | Hepatic Cirrhosis | • Can prolong QT |
| • The net effect is a concentration dependent prolongation of action potential duration and decreased in early after depolarizations. | |||||
| Ivabradine | • Ivabradine functions in a use-dependent fashion at the SA node inhibiting the mixed sodium-potassium current (If) thereby slowing depolarization of the pacemaker potential and lowering the heart rate | If | PO: 2.5–5 mg q12h | Bradycardia, heart block, sick sinus syndrome | • Symptomatic bradycardia, increase risk of atrial fibrillation |
| Adenosine | • Adenosine is an endogenous nucleoside that acts on adenosine receptors primarily located on the specialized conduction tissues of the SA and AV nodes resulting in activation of potassium channels and hyperpolarization and decreased automaticity of these tissues. | Activation of A1, A2, and IKATP | IV only: initial 6mg dose followed by 12 mg × 2 q1 minutes if peripherally administered. | Use in pre-excitation syndromes (Wolff-Parkinson-White) can precipitate VA’s. | • Can cause temporary, but profound, chest discomfort. |
| 50% dose reduction if via central line | |||||
| Arrhythmia Type | Etiology | Medication | Evidence |
|---|---|---|---|
| Brugada Syndrome | Brugada syndrome is an autosomal dominant genetic disorder with variable expression characterized by abnormal findings on the surface electrocardiogram (ECG) with an increased risk of ventricular tachyarrhythmias and sudden cardiac death. Most commonly, Brugada is the result of defective sodium channels leading the reduction of sodium inflow current and a subsequent reduction in the duration of action potentials. | First Line: Quinidine | Belhassen et al. [32] |
| Marquez et al. [31] | |||
| Long QT Syndrome | Long QT syndromes may be congenital or acquired and represent a disorder of myocardial repolarization characterized by a prolonged QT interval on the electrocardiogram (ECG). These findings lead to an increased risk of polymorphic VT, which can be life-threatening. This review focuses on congenital LQT in the structural normal heart. | ||
| Long QT1/Long QT2 | Both Long QT1 and Long QT2 are caused by mutations in genes encoding potassium channel leading to a defect in inward potassium current and QT prolongation. Their major difference is LQT1 effects KCNQ1 gene most commonly leading to a defect in slow potassium current (Iks) while LQT2 effects KCNH2 gene most commonly leading to a defect in rapid potassium current (Ikr). The overall effect and treatment remains the same for both types. | Beta Blockers (Propranolol/Nadolol preferred) | Bennett et al. [88] |
| Schwartz et al. [87] | |||
| Long QT3 | Unlike LQT1/LQT2, LQT3 is caused by a defect in SCN5A gene most commonly. This mutation leads to a defect in a cardiac sodium channel and subsequently increases the delayed Na+ inward current and, therefore, prolonging the action potential duration. As the primary effect is on sodium channels, it is amenable to treatments with mechanism of action on these channels. | First Line: Beta Blockers | Wilde et al. [89] |
| Second Line: Mexiletine | Mazzanti et al. [56] | ||
| Salvage Therapy: Flecainide/Propafenone | Moss et al. [63] | ||
| Belardinelli et al. [62] | |||
| Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) | CVPT is an often familial syndrome leading to exercise-induced polymorphic VT in childhood/adolescents. CVPT most commonly arises due to mutations in one of two genes: the cardiac ryanodine receptor gene (an autosomal dominant form) and the calsequestrin 2 gene (autosomal recessive). Both mutations act by inducing intracellular calcium release and causing a intracellular calcium overload. This overload leads to delayed afterdepolarization, which can induce ventricular arrhythmias. | First Line Beta Blockers (Nadolol with strongest evidence) | Priori et al. [86] |
| Second Line: Calcium Channel Blockers | Leenhardt et al. [85] | ||
| Rosso et al. [126] | |||
| Idiopathic Left Ventricular Tachycardia (ILVT) | ILVT is an idiopathic form a VT presenting in young adulthood of unclear etiology. Current studies indicate that this arrhythmia is caused by localized reentry circuit close to the posterior fascicle. | Calcium Channel Blockers | Belhassen et al. [121] |
| Ohe et al. [123] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larson, J.; Rich, L.; Deshmukh, A.; Judge, E.C.; Liang, J.J. Pharmacologic Management for Ventricular Arrhythmias: Overview of Anti-Arrhythmic Drugs. J. Clin. Med. 2022, 11, 3233. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11113233
Larson J, Rich L, Deshmukh A, Judge EC, Liang JJ. Pharmacologic Management for Ventricular Arrhythmias: Overview of Anti-Arrhythmic Drugs. Journal of Clinical Medicine. 2022; 11(11):3233. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11113233
Chicago/Turabian StyleLarson, John, Lucas Rich, Amrish Deshmukh, Erin C. Judge, and Jackson J. Liang. 2022. "Pharmacologic Management for Ventricular Arrhythmias: Overview of Anti-Arrhythmic Drugs" Journal of Clinical Medicine 11, no. 11: 3233. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11113233