
Sex/Gender- and Age-Related Differences in β-Adrenergic Receptor Signaling in Cardiovascular Diseases


{kind=link}
{kind=link}
Abstract
:1. Introduction
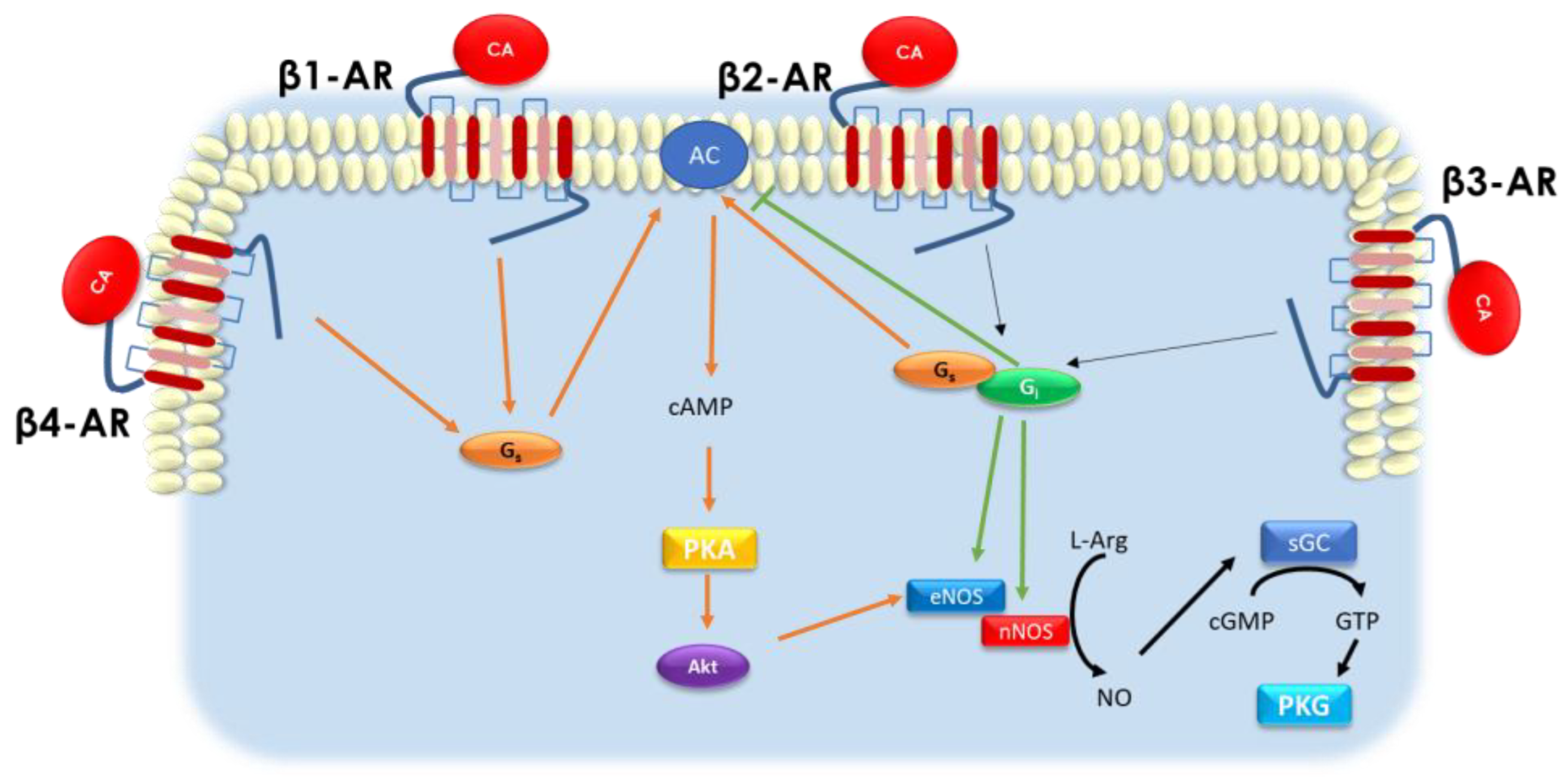
2. β-Adrenergic Receptors and Cardiovascular System
3. Targeting β-AR Signaling in Cardiovascular Disease: GRKs Inhibition, β-Blockade and Pharmacogenomics
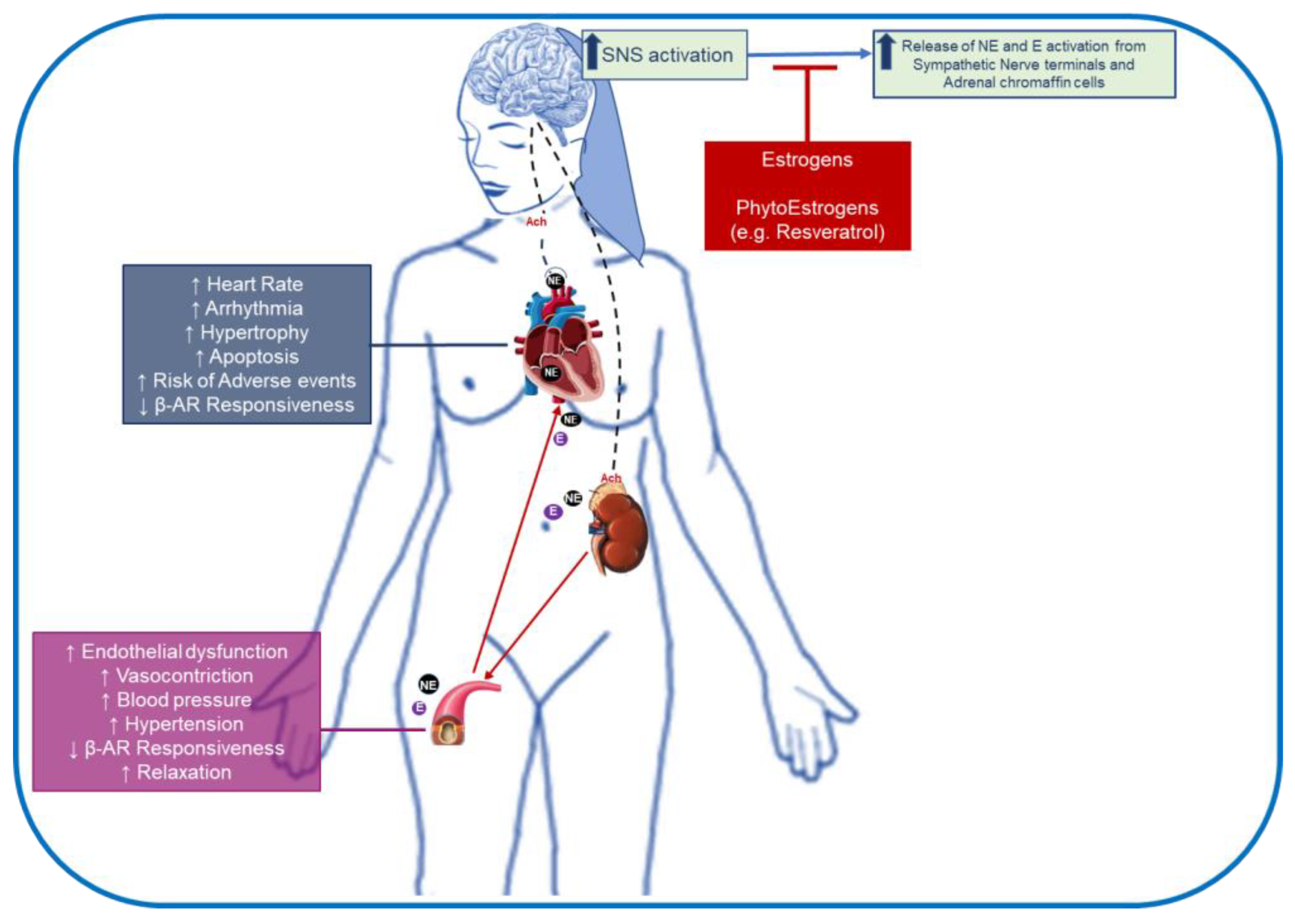
4. Sex- and Age-Related Differences in β-Adrenergic receptors: Impact on Cardiovascular Disease
5. Impact of Estrogens and Its Supplementation on β-AR Signaling: Implication in Cardiovascular Disease
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report from the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Lucà, F.; Abrignani, M.G.; Parrini, I.; Di Fusco, S.A.; Giubilato, S.; Rao, C.M.; Piccioni, L.; Cipolletta, L.; Passaretti, B.; Giallauria, F.; et al. Update on Management of Cardiovascular Diseases in Women. J. Clin. Med. 2022, 11, 1176. [Google Scholar] [CrossRef] [PubMed]
- Mendirichaga, R.; Jacobs, A.K. Sex Differences in Ischemic Heart Disease-the Paradox Persists. JAMA Cardiol. 2020, 5, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Timmis, A.; Townsend, N.; Gale, C.; Grobbee, R.; Maniadakis, N.; Flather, M.; Wilkins, E.; Wright, L.; Vos, R.; Bax, J.; et al. European Society of Cardiology: Cardiovascular Disease Statistics 2017. Eur. Heart J. 2018, 39, 508–579. [Google Scholar] [CrossRef] [Green Version]
- Ghare, M.I.; Chandrasekhar, J.; Mehran, R.; Ng, V.; Grines, C.; Lansky, A. Sex Disparities in Cardiovascular Device Evaluations: Strategies for Recruitment and Retention of Female Patients in Clinical Device Trials. JACC Cardiovasc. Interv. 2019, 12, 301–308. [Google Scholar] [CrossRef]
- Connelly, P.J.; Azizi, Z.; Alipour, P.; Delles, C.; Pilote, L.; Raparelli, V. The Importance of Gender to Understand Sex Differences in Cardiovascular Disease. Can. J. Cardiol. 2021, 37, 699–710. [Google Scholar] [CrossRef]
- Ueda, K.; Fukuma, N.; Adachi, Y.; Numata, G.; Tokiwa, H.; Toyoda, M.; Otani, A.; Hashimoto, M.; Liu, P.Y.; Takimoto, E. Sex Differences and Regulatory Actions of Estrogen in Cardiovascular System. Front. Physiol. 2021, 12, 738218. [Google Scholar] [CrossRef]
- Crescioli, C. The Role of Estrogens and Vitamin D in Cardiomyocyte Protection: A Female Perspective. Biomolecules 2021, 11, 1815. [Google Scholar] [CrossRef]
- Niță, A.R.; Knock, G.A.; Heads, R.J. Signalling mechanisms in the cardiovascular protective effects of estrogen: With a focus on rapid/membrane signalling. Curr. Res. Physiol. 2021, 4, 103–118. [Google Scholar] [CrossRef]
- Xu, S.; Xie, F.; Tian, L.; Fallah, S.; Babaei, F.; Manno, S.H.C.; Manno, F.A.M.; Zhu, L.; Wong, K.F.; Liang, Y.; et al. Estrogen accelerates heart regeneration by promoting the inflammatory response in zebrafish. J. Endocrinol. 2020, 245, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Crabbe, D.L.; Dipla, K.; Ambati, S.; Zafeiridis, A.; Gaughan, J.P.; Houser, S.R.; Margulies, K.B. Gender differences in post-infarction hypertrophy in end-stage failing hearts. J. Am. Coll. Cardiol. 2003, 41, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Barrett-Connor, E. Sex differences in coronary heart disease. Why are women so superior? The 1995 Ancel Keys Lecture. Circulation 1997, 95, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Hayward, C.S.; Kelly, R.P.; Collins, P. The roles of gender, the menopause and hormone replacement on cardiovascular function. Cardiovasc. Res. 2000, 46, 28–49. [Google Scholar] [CrossRef] [Green Version]
- Sabbatini, A.R.; Kararigas, G. Menopause-Related Estrogen Decrease and the Pathogenesis of HFpEF: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 75, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Hjortland, M.C.; McNamara, P.M.; Gordon, T. Menopause and risk of cardiovascular disease: The Framingham study. Ann. Intern. Med. 1976, 85, 447–452. [Google Scholar] [CrossRef]
- Steinberg, J.R.; Turner, B.E.; Weeks, B.T.; Magnani, C.J.; Wong, B.O.; Rodriguez, F.; Yee, L.M.; Cullen, M.R. Analysis of Female Enrollment and Participant Sex by Burden of Disease in US Clinical Trials between 2000 and 2020. JAMA Netw. Open. 2021, 4, e2113749. [Google Scholar] [CrossRef]
- Huang, Z.M.; Gold, J.I.; Koch, W.J. G protein-coupled receptor kinases in normal and failing myocardium. Front. Biosci. 2011, 16, 3047–3060. [Google Scholar] [CrossRef] [Green Version]
- Elia, A.; Cannavo, A.; Gambino, G.; Cimini, M.; Ferrara, N.; Kishore, R.; Paolocci, N.; Rengo, G. Aging is associated with cardiac autonomic nerve fiber depletion and reduced cardiac and circulating BDNF levels. J. Geriatr. Cardiol. 2021, 18, 549–559. [Google Scholar] [CrossRef]
- Bencivenga, L.; Palaia, M.E.; Sepe, I.; Gambino, G.; Komici, K.; Cannavo, A.; Femminella, G.D.; Rengo, G. Why Do We Not Assess Sympathetic Nervous System Activity in Heart Failure Management: Might GRK2 Serve as a New Biomarker? Cells 2021, 10, 457. [Google Scholar] [CrossRef]
- Cannavo, A.; Liccardo, D.; Koch, W.J. Targeting cardiac β-adrenergic signaling via GRK2 inhibition for heart failure therapy. Front Physiol. 2013, 4, 264. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, A.; Liccardo, D.; Lymperopoulos, A.; Santangelo, M.; Femminella, G.D.; Leosco, D.; Cittadini, A.; Ferrara, N.; Paolocci, N.; Koch, W.J.; et al. GRK2 Regulates α2-Adrenergic Receptor-Dependent Catecholamine Release in Human Adrenal Chromaffin Cells. J. Am. Coll. Cardiol. 2017, 69, 1515–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavo, A.; Koch, W.J. Targeting β3-Adrenergic Receptors in the Heart: Selective Agonism and β-Blockade. J. Cardiovasc. Pharmacol. 2017, 69, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavo, A.; Koch, W.J. GRK2 as negative modulator of NO bioavailability: Implications for cardiovascular disease. Cell Signal. 2018, 41, 33–40. [Google Scholar] [CrossRef]
- Motiejunaite, J.; Amar, L.; Vidal-Petiot, E. Adrenergic receptors and cardiovascular effects of catecholamines. Ann. Endocrinol. 2021, 82, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Flacco, N.; Segura, V.; Perez-Aso, M.; Estrada, S.; Seller, J.F.; Jiménez-Altayó, F.; Noguera, M.A.; D’Ocon, P.; Vila, E.; Ivorra, M.D. Different β-adrenoceptor subtypes coupling to cAMP or NO/cGMP pathways: Implications in the relaxant response of rat conductance and resistance vessels. Br. J. Pharmacol. 2013, 169, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Ufer, C.; Germack, R. Cross-regulation between beta 1- and beta 3-adrenoceptors following chronic beta-adrenergic stimulation in neonatal rat cardiomyocytes. Br. J. Pharmacol. 2009, 158, 300–313. [Google Scholar] [CrossRef] [Green Version]
- Germack, R.; Dickenson, J.M. Induction of beta3-adrenergic receptor functional expression following chronic stimulation with noradrenaline in neonatal rat cardiomyocytes. J. Pharmacol. Exp. Ther. 2006, 316, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Briones, A.M.; Daly, C.J.; Jimenez-Altayo, F.; Martinez-Revelles, S.; Gonzalez, J.M.; McGrath, J.C.; Vila, E. Direct demonstration of β1- and evidence against β2- and β3-adrenoceptors, in smooth muscle cells of rat small mesenteric arteries. Br. J. Pharmacol. 2005, 146, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.A.; Porter, K.E.; Smith, W.H.; White, H.L.; Ball, S.G.; Balmforth, A.J. Chronic beta2-adrenergic receptor stimulation increases proliferation of human cardiac fibroblasts via an autocrine mechanism. Cardiovasc. Res. 2003, 57, 784–792. [Google Scholar] [CrossRef] [Green Version]
- Wallukat, G. The beta-adrenergic receptors. Herz 2002, 27, 683–690. [Google Scholar] [CrossRef]
- Madamanchi, A. Beta-adrenergic receptor signaling in cardiac function and heart failure. McGill J. Med. 2007, 10, 99–104. [Google Scholar] [PubMed]
- Granneman, J.G. The putative beta4-adrenergic receptor is a novel state of the beta1-adrenergic receptor. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E199–E202. [Google Scholar] [CrossRef] [PubMed]
- Mohell, N.; Dicker, A. The β-adrenergic radioligand [3H]CGP-12177, generally classified as an antagonist, is a thermogenic agonist in brown adipose tissue. Biochem. J. 1989, 261, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Staehelin, M.; Simons, P.; Jaeggi, K.; Wigger, N. CGP-12177. A hydrophilic β-adrenergic receptor radioligand reveals high affinity binding of agonists to intact cells. J. Biol. Chem. 1983, 258, 3496–3502. [Google Scholar] [CrossRef]
- Ito, M.; Grujic, D.; Abel, E.D.; Vidal-Puig, A.; Susulic, V.S.; Lawitts, J.; Harper, M.E.; Himms-Hagen, J.; Strosberg, A.D.; Lowell, B.B. Mice expressing human but not murine beta3-adrenergic receptors under the control of human gene regulatory elements. Diabetes 1998, 47, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Preitner, F.; Muzzin, P.; Revelli, J.P.; Seydoux, J.; Galitzky, J.; Berlan, M.; Lafontan, M.; Giacobino, J.P. Metabolic response to various beta-adrenoceptor agonists in β3-adrenoceptor knockout mice: Evidence for a new β-adrenergic receptor in brown adipose tissue. Br. J. Pharmacol. 1998, 124, 1684–1688. [Google Scholar] [CrossRef] [Green Version]
- Galitzky, J.; Langin, D.; Verwaerde, P.; Montastruc, J.L.; Lafontan, M.; Berlan, M. Lipolytic effects of conventional β3-adrenoceptor agonists and of CGP 12, 177 in rat and human fat cells: Preliminary pharmacological evidence for a putative β4-adrenoceptor. Br. J. Pharmacol. 1997, 122, 1244–1250. [Google Scholar] [CrossRef] [Green Version]
- Kaumann, A.J.; Molenaar, P. Differences between the third cardiac β-adrenoceptor and the colonic β3-adrenoceptor in the rat. Br. J. Pharmacol. 1996, 118, 2085–2098. [Google Scholar] [CrossRef]
- Kaumann, A.J.; Molenaar, P. Modulation of human cardiac function through 4 β-adrenoceptor populations. Naunyn-Schmiedebergs Arch. Pharmacol. 1997, 355, 667–681. [Google Scholar] [CrossRef]
- Kaumann, A.J.; Preitner, F.; Sarsero, D.; Molenaar, P.; Revelli, J.P.; Giacobino, J.P. (−)-CGP 12177 causes cardiostimulation and binds to cardiac putative β4-adrenoceptors in both wild-type and β3-adrenoceptor knockout mice. Mol. Pharmacol. 1998, 53, 670–675. [Google Scholar] [CrossRef]
- Cannavo, A. G Protein-Coupled Receptor and Their Kinases in Cell Biology and Disease. Int. J. Mol. Sci. 2022, 23, 5501. [Google Scholar] [CrossRef] [PubMed]
- Wettschureck, N.; Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trappanese, D.M.; Liu, Y.; McCormick, R.C.; Cannavo, A.; Nanayakkara, G.; Baskharoun, M.M.; Jarrett, H.; Woitek, F.J.; Tillson, D.M.; Dillon, A.R.; et al. Chronic β1-adrenergic blockade enhances myocardial β3-adrenergic coupling with nitric oxide-cGMP signaling in a canine model of chronic volume overload: New insight into mechanisms of cardiac benefit with selective β1-blocker therapy. Basic Res. Cardiol. 2015, 110, 456. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, E. Cyclic GMP-dependent signaling in cardiac myocytes. Circ. J. 2012, 76, 1819–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvert, J.W.; Condit, M.E.; Aragón, J.P.; Nicholson, C.K.; Moody, B.F.; Hood, R.L.; Sindler, A.L.; Gundewar, S.; Seals, D.R.; Barouch, L.A.; et al. Exercise protects against myocardial ischemia-reperfusion injury via stimulation of β(3)-adrenergic receptors and increased nitric oxide signaling: Role of nitrite and nitrosothiols. Circ. Res. 2011, 108, 1448–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queen, L.R.; Ji, Y.; Xu, B.; Young, L.; Yao, K.; Wyatt, A.W.; Rowlands, D.J.; Siow, R.C.; Mann, G.E.; Ferro, A. Mechanisms underlying beta2-adrenoceptor-mediated nitric oxide generation by human umbilical vein endothelial cells. J. Physiol. 2006, 576, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Ferro, A.; Coash, M.; Yamamoto, T.; Rob, J.; Ji, Y.; Queen, L. Nitric oxide-dependent beta2-adrenergic dilatation of rat aorta is mediated through activation of both protein kinase A and Akt. Br. J. Pharmacol. 2004, 143, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, A.; Liccardo, D.; Lymperopoulos, A.; Gambino, G.; D’Amico, M.L.; Rengo, F.; Koch, W.J.; Leosco, D.; Ferrara, N.; Rengo, G. β Adrenergic Receptor Kinase C-Terminal Peptide Gene-Therapy Improves β2-Adrenergic Receptor-Dependent Neoangiogenesis after Hindlimb Ischemia. J. Pharmacol. Exp. Ther. 2016, 356, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Tanner, M.A.; Thomas, T.P.; Maitz, C.A.; Grisanti, L.A. β2-Adrenergic Receptors Increase Cardiac Fibroblast Proliferation Through the Gαs/ERK1/2-Dependent Secretion of Interleukin-6. Int. J. Mol. Sci. 2020, 21, 8507. [Google Scholar] [CrossRef]
- Gauthier, C.; Leblais, V.; Kobzik, L.; Trochu, J.N.; Khandoudi, N.; Bril, A.; Balligand, J.L.; Le Marec, H. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J. Clin. Investig. 1998, 102, 1377–1384. [Google Scholar] [CrossRef]
- Gauthier, C.; Tavernier, G.; Charpentier, F.; Langin, D.; Le Marec, H. Functional beta3-adrenoceptor in the human heart. J. Clin. Investig. 1996, 98, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Communal, C.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis: Role of a pertussis toxin-sensitive G protein. Circulation 1999, 100, 2210–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaugg, M.; Xu, W.; Lucchinetti, E.; Shafiq, S.A.; Jamali, N.Z.; Siddiqui, M.A. Beta-adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation 2000, 102, 344–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, M.; Frischkopf, K.; Taimor, G.; Piper, H.M.; Schluter, K.D. Hypertrophic effect of selective β1-adrenoceptor stimulation on ventricular cardiomyocytes from adult rat. Am. J. Physiol. Cell Physiol. 2000, 279, C495–C503. [Google Scholar] [CrossRef]
- Morisco, C.; Zebrowski, D.C.; Vatner, D.E.; Vatner, S.F.; Sadoshima, J. β-adrenergic cardiac hypertrophy is mediated primarilyby the β1-subtype in the rat heart. J. Mol. Cell. Cardiol. 2001, 33, 561–573. [Google Scholar] [CrossRef]
- Tobin, A.B. G-protein-coupled receptor phosphorylation: Where, when and by whom. Br. J. Pharmacol. 2008, 153, S167–S176. [Google Scholar] [CrossRef] [Green Version]
- Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 1997, 390, 88–91. [Google Scholar] [CrossRef]
- Echeverría, E.; Cabrera, M.; Burghi, V.; Sosa, M.; Ripoll, S.; Yaneff, A.; Monczor, F.; Davio, C.; Shayo, C.; Fernández, N. The Regulator of G Protein Signaling Homologous Domain of G Protein-Coupled Receptor Kinase 2 Mediates Short-Term Desensitization of β3-Adrenergic Receptor. Front. Pharmacol. 2020, 11, 113. [Google Scholar] [CrossRef]
- Cannavo, A.; Komici, K.; Bencivenga, L.; D’Amico, M.L.; Gambino, G.; Liccardo, D.; Ferrara, N.; Rengo, G. GRK2 as a therapeutic target for heart failure. Expert Opin. Ther. Targets 2018, 22, 75–83. [Google Scholar] [CrossRef]
- Marzano, F.; Rapacciuolo, A.; Ferrara, N.; Rengo, G.; Koch, W.J.; Cannavo, A. Targeting GRK5 for Treating Chronic Degenerative Diseases. Int. J. Mol. Sci. 2021, 22, 1920. [Google Scholar] [CrossRef]
- Rengo, G.; Pagano, G.; Paolillo, S.; de Lucia, C.; Femminella, G.D.; Liccardo, D.; Cannavo, A.; Formisano, R.; Petraglia, L.; Komici, K.; et al. Impact of diabetes mellitus on lymphocyte GRK2 protein levels in patients with heart failure. Eur. J. Clin. Investig. 2015, 45, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavo, A.; Rengo, G.; Liccardo, D.; Pagano, G.; Zincarelli, C.; De Angelis, M.C.; Puglia, R.; Di Pietro, E.; Rabinowitz, J.E.; Barone, M.V.; et al. β1-adrenergic receptor and sphingosine-1-phosphate receptor 1 (S1PR1) reciprocal downregulation influences cardiac hypertrophic response and progression to heart failure: Protective role of S1PR1 cardiac gene therapy. Circulation 2013, 128, 1612–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavo, A.; Marzano, F.; Elia, A.; Liccardo, D.; Bencivenga, L.; Gambino, G.; Perna, C.; Rapacciuolo, A.; Cittadini, A.; Ferrara, N.; et al. Aldosterone Jeopardizes Myocardial Insulin and β-Adrenergic Receptor Signaling via G Protein-Coupled Receptor Kinase 2. Front. Pharmacol. 2019, 10, 888. [Google Scholar] [CrossRef] [Green Version]
- Rengo, G.; Parisi, V.; Femminella, G.D.; Pagano, G.; de Lucia, C.; Cannavo, A.; Liccardo, D.; Giallauria, F.; Scala, O.; Zincarelli, C.; et al. Molecular aspects of the cardioprotective effect of exercise in the elderly. Aging Clin. Exp. Res. 2013, 25, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Nash, C.A.; Nelson, C.P.; Mistry, R.; Moeller-Olsen, C.; Christofidou, E.; Challiss, R.A.J.; Willets, J.M. Differential regulation of β2-adrenoceptor and adenosine A2B receptor signalling by GRK and arrestin proteins in arterial smooth muscle. Cell Signal. 2018, 51, 86–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hullmann, J.; Traynham, C.J.; Coleman, R.C.; Koch, W.J. The expanding GRK interactome: Implications in cardiovascular disease and potential for therapeutic development. Pharmacol. Res. 2016, 110, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzano, F.; Liccardo, D.; Elia, A.; Mucio, I.; de Lucia, C.; Lucchese, A.M.; Gao, E.; Ferrara, N.; Rapacciuolo, A.; Paolocci, N.; et al. Genetic Catalytic Inactivation of GRK5 Impairs Cardiac Function in Mice Via Dysregulated P53 Levels. JACC Basic Transl. Sci. 2022, 7, 366–380. [Google Scholar] [CrossRef]
- Ikeda, S.; Kaneko, M.; Fujiwara, S. Cardiotonic Agent Comprising GRK Inhibitor. Patent WO2007034846, 29 March 2007. [Google Scholar]
- Thal, D.M.; Homan, K.T.; Chen, J.; Wu, E.K.; Hinkle, P.M.; Huang, Z.M.; Chuprun, J.K.; Song, J.; Gao, E.; Cheung, J.Y.; et al. Paroxetine is a direct inhibitor of g protein-coupled receptor kinase 2 and increases myocardial contractility. ACS Chem. Biol. 2012, 7, 1830–1839. [Google Scholar] [CrossRef]
- Bouley, R.; Waldschmidt, H.V.; Cato, M.C.; Cannavo, A.; Song, J.; Cheung, J.Y.; Yao, X.Q.; Koch, W.J.; Larsen, S.D.; Tesmer, J.J.G. Structural Determinants Influencing the Potency and Selectivity of Indazole-Paroxetine Hybrid G Protein-Coupled Receptor Kinase 2 Inhibitors. Mol. Pharmacol. 2017, 92, 707–717. [Google Scholar] [CrossRef]
- Waldschmidt, H.V.; Homan, K.T.; Cato, M.C.; Cruz-Rodríguez, O.; Cannavo, A.; Wilson, M.W.; Song, J.; Cheung, J.Y.; Koch, W.J.; Tesmer, J.J.; et al. Structure-Based Design of Highly Selective and Potent G Protein-Coupled Receptor Kinase 2 Inhibitors Based on Paroxetine. J. Med. Chem. 2017, 60, 3052–3069. [Google Scholar] [CrossRef] [Green Version]
- Homan, K.T.; Wu, E.; Cannavo, A.; Koch, W.J.; Tesmer, J.J. Identification and characterization of amlexanox as a G protein-coupled receptor kinase 5 inhibitor. Molecules 2014, 19, 16937–16949. [Google Scholar] [CrossRef] [Green Version]
- Winstel, R.; Ihlenfeldt, H.G.; Jung, G.; Krasel, C.; Lohse, M.J. Peptide inhibitors of G protein-coupled receptorkinases. Biochem. Pharmacol. 2005, 70, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Doronin, S.; Lin, F.; Wang, H.Y.; Malbon, C.C. The full-length, cytoplasmic C-terminus of the beta 2-adrenergic receptor expressed in E. coli acts as a substrate for phosphorylation by protein kinase A, insulin receptor tyrosine kinase, GRK2, but not protein kinase C and suppresses desensitization when expressed in vivo. Protein Expr. Purif. 2000, 20, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Koch, W.J.; Inglese, J.; Stone, W.C.; Lefkowitz, R.J. The binding site for the beta gamma subunits of heterotrimeric G proteins on the beta-adrenergic receptor kinase. J. Biol. Chem. 1993, 268, 8256–8260. [Google Scholar] [CrossRef]
- Koch, W.J.; Rockman, H.A.; Samama, P.; Hamilton, R.A.; Bond, R.A.; Milano, C.A.; Lefkowitz, R.J. Cardiac function in mice overexpressing the beta-adrenergic receptor kinase or a beta ARK inhibitor. Science 1995, 268, 1350–1353. [Google Scholar] [CrossRef]
- Mayor, F., Jr.; Lucas, E.; Jurado-Pueyo, M.; Garcia-Guerra, L.; Nieto-Vazquez, I.; Vila-Bedmar, R.; Fernández-Veledo, S.; Murga, C. G Protein-coupled receptor kinase 2 (GRK2): A novel modulator of insulin resistance. Arch. Physiol. Biochem. 2011, 117, 125–130. [Google Scholar] [CrossRef]
- Cannavo, A.; Liccardo, D.; Eguchi, A.; Elliott, K.J.; Traynham, C.J.; Ibetti, J.; Eguchi, S.; Leosco, D.; Ferrara, N.; Rengo, G.; et al. Myocardial pathology induced by aldosterone is dependent on non-canonical activities of G protein-coupled receptor kinases. Nat. Commun. 2016, 7, 10877. [Google Scholar] [CrossRef] [Green Version]
- Oliver, E.; Mayor, F., Jr.; D’Ocon, P. Beta-blockers: Historical Perspective and Mechanisms of Action. Rev. Esp. Cardiol. 2019, 72, 853–862. [Google Scholar] [CrossRef]
- Dézsi, C.A.; Szentes, V. The Real Role of β-Blockers in Daily Cardiovascular Therapy. Am. J. Cardiovasc. Drugs 2017, 17, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Silverio, A.; Parodi, G.; Scudiero, F.; Bossone, E.; Di Maio, M.; Vriz, O.; Bellino, M.; Zito, C.; Provenza, G.; Radano, I.; et al. Beta-blockers are associated with better long-term survival in patients with Takotsubo syndrome. Heart 2022, 108, 1244–1245. [Google Scholar] [CrossRef]
- Iaccarino, G.; Tomhave, E.D.; Lefkowitz, R.J.; Koch, W.J. Reciprocal in vivo regulation of myocardial G protein-coupled receptor kinase expression by beta-adrenergic receptor stimulation and blockade. Circulation 1998, 98, 1783–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rengo, G.; Lymperopoulos, A.; Zincarelli, C.; Donniacuo, M.; Soltys, S.; Rabinowitz, J.E.; Koch, W.J. Myocardial adeno-associated virus serotype 6-betaARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation 2009, 119, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leineweber, K.; Rohe, P.; Beilfuss, A.; Wolf, C.; Sporkmann, H.; Bruck, H.; Jakob, H.G.; Heusch, G.; Philipp, T.; Brodde, O.E. G-protein-coupled receptor kinase activity in human heart failure: Effects of beta-adrenoceptor blockade. Cardiovasc. Res. 2005, 66, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, J.G.; Hill, S.J.; Summers, R.J. Evolution of β-blockers: From anti-anginal drugs to ligand-directed signalling. Trends Pharmacol. Sci. 2011, 32, 227–234. [Google Scholar] [CrossRef]
- Cannavo, A.; Rengo, G.; Liccardo, D.; Pun, A.; Gao, E.; George, A.J.; Gambino, G.; Rapacciuolo, A.; Leosco, D.; Ibanez, B.; et al. β1-Blockade Prevents Post-Ischemic Myocardial Decompensation Via β3AR-Dependent Protective Sphingosine-1 Phosphate Signaling. J. Am. Coll. Cardiol. 2017, 70, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Parsons, H.; Allard, M.F.; McNeill, J.H. Metoprolol increases the expression of β3-adrenoceptors in the diabetic heart: Effects on nitric oxide signaling and forkhead transcription factor-3. Eur. J. Pharmacol. 2008, 595, 44–51. [Google Scholar] [CrossRef]
- Aragón, J.P.; Condit, M.E.; Bhushan, S.; Predmore, B.L.; Patel, S.S.; Grinsfelder, D.B.; Gundewar, S.; Jha, S.; Calvert, J.W.; Barouch, L.A.; et al. Beta3-adrenoreceptor stimulation ameliorates myocardial ischemia-reperfusion injury via endothelial nitric oxide synthase and neuronal nitric oxide synthase activation. J. Am. Coll. Cardiol. 2011, 58, 2683–2691. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Ding, L.; Jin, Z.; Gao, G.; Li, H.; Zhang, L.; Zhang, L.; Lu, X.; Hu, L.; Lu, B.; et al. Nebivolol protects against myocardial infarction injury via stimulation of beta 3-adrenergic receptors and nitric oxide signaling. PLoS ONE 2014, 9, e98179. [Google Scholar] [CrossRef]
- Dessy, C.; Saliez, J.; Ghisdal, P.; Daneau, G.; Lobysheva, I.I.; Frérart, F.; Belge, C.; Jnaoui, K.; Noirhomme, P.; Feron, O.; et al. Endothelial beta3-adrenoreceptors mediate nitric oxide-dependent vasorelaxation of coronary microvessels in response to the third-generation beta-blocker nebivolol. Circulation 2005, 112, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, S.A.; Doerries, C.; Manes, C.; Speer, T.; Dessy, C.; Lobysheva, I.; Mohmand, W.; Akbar, R.; Bahlmann, F.; Besler, C.; et al. Nebivolol exerts beneficial effects on endothelial function, early endothelial progenitor cells, myocardial neovascularization, and left ventricular dysfunction early after myocardial infarction beyond conventional β1-blockade. J. Am. Coll. Cardiol. 2011, 57, 601–611. [Google Scholar] [CrossRef] [Green Version]
- Rengo, G.; Cannavo, A.; Liccardo, D.; Zincarelli, C.; de Lucia, C.; Pagano, G.; Komici, K.; Parisi, V.; Scala, O.; Agresta, A.; et al. Vascular endothelial growth factor blockade prevents the beneficial effects of β-blocker therapy on cardiac function, angiogenesis, and remodeling in heart failure. Circ. Heart Fail. 2013, 6, 1259–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakoki, M.; Hirata, Y.; Hayakawa, H.; Nishimatsu, H.; Suzuki, Y.; Nagata, D.; Suzuki, E.; Kikuchi, K.; Nagano, T.; Omata, M. Effects of vasodilatory β-adrenoceptor antagonists on endothelium-derived nitric oxide release in rat kidney. Hypertension 1999, 33, 467–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhang, Q.; Zhang, H.; Wang, C.; Xiu, R. Effects of nebivolol versus other antihypertensive drugs on the endothelial dysfunction in patients with essential hypertension. Biosci. Rep. 2020, 40, BSR20200436. [Google Scholar] [CrossRef]
- do Vale, G.T.; Simplicio, J.A.; Gonzaga, N.A.; Yokota, R.; Ribeiro, A.A.; Casarini, D.E.; de Martinis, B.S.; Tirapelli, C.R. Nebivolol prevents vascular oxidative stress and hypertension in rats chronically treated with ethanol. Atherosclerosis 2018, 274, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Current role of beta-adrenergic blockers in the management of chronic heart failure. Am. J. Med. 2001, 110, 81S–94S. [Google Scholar] [CrossRef]
- Koch, W.J.; Cannavo, A. Eating Away at Heart Failure. J. Am. Coll. Cardiol. 2015, 66, 2534–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leineweber, K.; Heusch, G. Beta 1- and beta 2-adrenoceptor polymorphisms and cardiovascular diseases. Br. J. Pharmacol. 2009, 158, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Liggett, S.B. Pharmacogenomics of beta1-adrenergic receptor polymorphisms in heart failure. Heart Fail Clin. 2010, 6, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Mialet Perez, J.; Rathz, D.A.; Petrashevskaya, N.N.; Hahn, H.S.; Wagoner, L.E.; Schwartz, A.; Dorn, G.W.; Liggett, S.B. Beta 1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat. Med. 2003, 9, 1300–1305. [Google Scholar] [CrossRef]
- Liggett, S.B.; Mialet-Perez, J.; Thaneemit-Chen, S.; Weber, S.A.; Greene, S.M.; Hodne, D.; Nelson, B.; Morrison, J.; Domanski, M.J.; Wagoner, L.E.; et al. A polymorphism within a conserved beta(1)-adrenergic receptor motif alters cardiac function and beta-blocker response in human heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 11288–11293. [Google Scholar] [CrossRef] [Green Version]
- Luzum, J.A.; English, J.D.; Ahmad, U.S.; Sun, J.W.; Canan, B.D.; Sadee, W.; Kitzmiller, J.P.; Binkley, P.F. Association of Genetic Polymorphisms in the Beta-1 Adrenergic Receptor with Recovery of Left Ventricular Ejection Fraction in Patients with Heart Failure. J. Cardiovasc. Transl. Res. 2019, 12, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, C.; Song, Y.; Fan, X.; You, L.; Tan, L.; Xiao, L.; Li, Q.; Ruan, G.; Hu, S.; et al. ADRB2 polymorphism Arg16Gly modifies the natural outcome of heart failure and dictates therapeutic response to β-blockers in patients with heart failure. Cell Discov. 2018, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Liggett, S.B.; Cresci, S.; Kelly, R.J.; Syed, F.M.; Matkovich, S.J.; Hahn, H.S.; Diwan, A.; Martini, J.S.; Sparks, L.; Parekh, R.R.; et al. A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat. Med. 2008, 14, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.; Radhakrishnan, S.; Kaliappan, T.; Gopalan, R.; Subrahmanian, M.; Sankaran, R. The genetics of cardiac failure: Role of a G protein-coupled receptor polymorphism in therapeutic response in an Indian population. J. Clin. Transl. Res. 2021, 7, 501–510. [Google Scholar] [PubMed]
- Kang, S.; Hong, X.; Ruan, C.W.; Yu, P.; Yu, S.S.; Chen, M.; Zhang, D.F.; Fan, H.M.; Liu, Z.M. Effects of GRK5 and ADRB1 polymorphisms influence on systolic heart failure. J. Transl. Med. 2015, 13, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurnik, D.; Cunningham, A.J.; Sofowora, G.G.; Kohli, U.; Li, C.; Friedman, E.A.; Muszkat, M.; Menon, U.B.; Wood, A.J.; Stein, C.M. GRK5 Gln41Leu polymorphism is not associated with sensitivity to beta(1)-adrenergic blockade in humans. Pharmacogenomics 2009, 10, 1581–1587. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.E.; Hanley-Yanez, K.; Yancy, C.W.; Taylor, A.L.; Feldman, A.M.; McNamara, D.M. Adrenergic Polymorphisms and Survival in African Americans with Heart Failure: Results From A-HeFT. J. Card. Fail. 2019, 25, 553–560. [Google Scholar] [CrossRef]
- de Groote, P.; Helbecque, N.; Lamblin, N.; Hermant, X.; Mc Fadden, E.; Foucher-Hossein, C.; Amouyel, P.; Dallongeville, J.; Bauters, C. Association between beta-1 and beta-2 adrenergic receptor gene polymorphisms and the response to beta-blockade in patients with stable congestive heart failure. Pharmacogenet. Genomics. 2005, 15, 137–142. [Google Scholar] [CrossRef]
- Santema, B.T.; Ouwerkerk, W.; Tromp, J.; Sama, I.E.; Ravera, A.; Regitz-Zagrosek, V.; Hillege, H.; Samani, N.J.; Zannad, F.; Dickstein, K.; et al. Identifying optimal doses of heart failure medications in men compared with women: A prospective, observational, cohort study. Lancet 2019, 394, 1254–1263. [Google Scholar] [CrossRef] [Green Version]
- Labbé, L.; Sirois, C.; Pilote, S.; Arseneault, M.; Robitaille, N.M.; Turgeon, J.; Hamelin, B.A. Effect of gender, sex hormones, time variables and physiological urinary pH on apparent CYP2D6 activity as assessed by metabolic ratios of marker substrates. Pharmacogenetics 2000, 10, 425–438. [Google Scholar] [CrossRef]
- Tanaka, E.; Hisawa, S. Clinically significant pharmacokinetic drug interactions with psychoactive drugs: Antidepressants and antipsychotics and the cytochrome P450 system. J. Clin. Pharm. Ther. 1999, 24, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Luzier, A.B.; Killian, A.; Wilton, J.H.; Wilson, M.F.; Forrest, A.; Kazierad, D.J. Gender-related effects on metoprolol pharmacokinetics and pharmacodynamics in healthy volunteers. Clin. Pharmacol. Ther. 1999, 66, 594–601. [Google Scholar] [CrossRef]
- Jochmann, N.; Stangl, K.; Garbe, E.; Baumann, G.; Stangl, V. Female-specific aspects in the pharmacotherapy of chronic cardiovascular diseases. Eur. Heart J. 2005, 26, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Soldin, O.P.; Mattison, D.R. Sex differences in pharmacokinetics and pharmacodynamics. Clin. Pharmacokinet. 2009, 48, 143–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitley, H.; Lindsey, W. Sex-based differences in drug activity. Am. Fam. Physician. 2009, 80, 1254–1258. [Google Scholar] [PubMed]
- Eugene, A.R. Gender based Dosing of Metoprolol in the Elderly using Population Pharmacokinetic Modeling and Simulations. Int. J. Clin. Pharmacol. Toxicol. 2016, 5, 209–215. [Google Scholar] [PubMed]
- Ueno, K.; Sato, H. Sex-related differences in pharmacokinetics and pharmacodynamics of anti-hypertensive drugs. Hypertens Res. 2012, 35, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2021 Update: A Report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Kneale, B.J.; Chowienczyk, P.J.; Brett, S.E.; Coltart, D.J.; Ritter, J.M. Gender differences in sensitivity to adrenergic agonists of forearm resistance vasculature. J. Am. Coll. Cardiol. 2000, 36, 1233–1238. [Google Scholar] [CrossRef] [Green Version]
- Al-Gburi, S.; Deussen, A.; Zatschler, B.; Weber, S.; Künzel, S.; El-Armouche, A.; Lorenz, K.; Cybularz, M.; Morawietz, H.; Kopaliani, I. Sex-difference in expression and function of beta-adrenoceptors in macrovessels: Role of the endothelium. Basic Res. Cardiol. 2017, 112, 29. [Google Scholar] [CrossRef]
- Riedel, K.; Deussen, A.J.; Tolkmitt, J.; Weber, S.; Schlinkert, P.; Zatschler, B.; Friebel, C.; Müller, B.; El-Armouche, A.; Morawietz, H.; et al. Estrogen determines sex differences in adrenergic vessel tone by regulation of endothelial β-adrenoceptor expression. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H243–H254. [Google Scholar] [CrossRef] [PubMed]
- Dart, A.M.; Du, X.J.; Kingwell, B.A. Gender, sex hormones and autonomic nervous control of the cardiovascular system. Cardiovasc. Res 2002, 53, 678–687. [Google Scholar] [CrossRef]
- Freedman, R.R.; Sabharwal, S.C.; Desai, N. Sex differences in peripheral vascular adrenergic receptors. Circ. Res. 1987, 61, 581–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.M.; Gzik, D.J. Sympatholytic interventions and vascular remodelling. Basic Res. Cardiol. 1991, 86, 55–64. [Google Scholar] [PubMed]
- Loria, A.S.; Brinson, K.N.; Fox, B.M.; Sullivan, J.C. Sex-specific alterations in NOS regulation of vascular function in aorta and mesenteric arteries from spontaneously hypertensive rats compared to Wistar Kyoto rats. Physiol. Rep. 2014, 2, e12125. [Google Scholar] [CrossRef] [Green Version]
- Hart, E.C.; Charkoudian, N.; Wallin, B.G.; Curry, T.B.; Eisenach, J.; Joyner, M.J. Sex and ageing differences in resting arterial pressure regulation: The role of the β-adrenergic receptors. J. Physiol. 2011, 589, 5285–5297. [Google Scholar] [CrossRef]
- Baker, S.E.; Limberg, J.K.; Ranadive, S.M.; Joyner, M.J. Neurovascular control of blood pressure is influenced by aging, sex, and sex hormones. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R1271–R1275. [Google Scholar] [CrossRef]
- Vizgirda, V.M.; Wahler, G.M.; Sondgeroth, K.L.; Ziolo, M.T.; Schwertz, D.W. Mechanisms of sex differences in rat cardiac myocyte response to β-adrenergic stimulation. Am. J. Physiology-Heart Circ. Physiol. 2002, 282, H256–H263. [Google Scholar] [CrossRef] [Green Version]
- Schwertz, D.W.; Vizgirda, V.; Solaro, R.J.; Piano, M.R.; Ryjewski, C. Sexual dimorphism in rat left atrial function and response to adrenergic stimulation. Mol. Cell. Biochem. 1999, 200, 143–153. [Google Scholar] [CrossRef]
- Curl, C.L.; Wendt, I.R.; Kotsanas, G. Effects of gender on intracellular [Ca2+] in rat cardiac myocytes. Pflugers. Arch Eur. J. Physiol. 2001, 441, 709–716. [Google Scholar] [CrossRef]
- Peter, A.K.; Walker, C.J.; Ceccato, T.; Trexler, C.L.; Ozeroff, C.D.; Lugo, K.R.; Perry, A.R.; Anseth, K.S.; Leinwand, L.A. Cardiac Fibroblasts Mediate a Sexually Dimorphic Fibrotic Response to β-Adrenergic Stimulation. J. Am. Heart Assoc. 2021, 10, e018876. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, A.; Park, S.B.; Hughes, J.W.; Blumenthal, J.A.; Hinderliter, A.; Trivedi, R.; McFetridge-Durdle, J. Cardiovascular hemodynamics during stress in premenopausal versus postmenopausal women. Menopause 2010, 17, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rengo, G.; Pagano, G.; Vitale, D.F.; Formisano, R.; Komici, K.; Petraglia, L.; Parisi, V.; Femminella, G.D.; de Lucia, C.; Paolillo, S.; et al. Impact of aging on cardiac sympathetic innervation measured by 123I-mIBG imaging in patients with systolic heart failure. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Navarro, R. Takotsubo syndrome: The broken-heart syndrome. Br. J. Cardiol. 2021, 28, 30–34. [Google Scholar] [CrossRef]
- Minhas, A.S.; Hughey, A.B.; Kolias, T.J. Nationwide Trends in Reported Incidence of Takotsubo Cardiomyopathy from 2006 to 2012. Am. J. Cardiol. 2015, 116, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Yang, G.; Kowitz, J.; Akin, I.; Zhou, X.; El-Battrawy, I. Takotsubo Syndrome: Translational Implications and Pathomechanisms. Int. J. Mol. Sci. 2022, 23, 1951. [Google Scholar] [CrossRef]
- Ali, A.; Redfors, B.; Lundgren, J.; Alkhoury, J.; Oras, J.; Gan, L.M.; Omerovic, E. Effects of pretreatment with cardiostimulants and beta-blockers on isoprenaline-induced takotsubo-like cardiac dysfunction in rats. Int. J. Cardiol. 2019, 281, 99–104. [Google Scholar] [CrossRef]
- Santoro, F.; Ieva, R.; Musaico, F.; Ferraretti, A.; Triggiani, G.; Tarantino, N.; Di Biase, M.; Brunetti, N.D. Lack of efficacy of drug therapy in preventing takotsubo cardiomyopathy recurrence: A meta-analysis. Clin. Cardiol. 2014, 37, 434–439. [Google Scholar] [CrossRef]
- Akashi, Y.J.; Goldstein, D.S.; Barbaro, G.; Ueyama, T. Takotsubo cardiomyopathy: A new form of acute, reversible heart failure. Circulation 2008, 118, 2754–2762. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, T. Takotsubo cardiomyopathy, a new concept of cardiomyopathy: Clinical features and pathophysiology. Int. J. Cardiol. 2015, 182, 297–303. [Google Scholar] [CrossRef]
- Isogai, T.; Matsui, H.; Tanaka, H.; Fushimi, K.; Yasunaga, H. Early beta-blocker use and in-hospital mortality in patients with Takotsubo cardiomyopathy. Heart 2016, 102, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Lerman, A.; Rihal, C.S. Apical ballooning syndrome (Tako-Tsubo or stress cardiomyopathy): A mimic of acute myocardial infarction. Am. Heart J. 2008, 155, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Kurisu, S.; Kihara, Y. Clinical management of takotsubo cardiomyopathy. Circ. J. 2014, 78, 1559–1566. [Google Scholar] [CrossRef] [Green Version]
- Evison, I.; Watson, G.; Chan, C.; Bridgman, P. The effects of beta-blockers in patients with stress cardiomyopathy. Intern. Med. J. 2021, 51, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Onoue, K.; Nakada, Y.; Nakagawa, H.; Kumazawa, T.; Ueda, T.; Nishida, T.; Soeda, T.; Okayama, S.; Watanabe, M.; et al. Alteration of β-Adrenoceptor Signaling in Left Ventricle of Acute Phase Takotsubo Syndrome: A Human Study. Sci Rep. 2018, 8, 12731. [Google Scholar] [CrossRef] [PubMed]
- Arcones, A.C.; Martínez-Cignoni, M.R.; Vila-Bedmar, R.; Yáñez, C.; Lladó, I.; Proenza, A.M.; Mayor, F., Jr.; Murga, C. Cardiac GRK2 Protein Levels Show Sexual Dimorphism during Aging and Are Regulated by Ovarian Hormones. Cells 2021, 10, 673. [Google Scholar] [CrossRef]
- Lindenfeld, J.; Cleveland, J.C., Jr.; Kao, D.P.; White, M.; Wichman, S.; Bristow, J.C.; Peterson, V.; Rodegheri-Brito, J.; Korst, A.; Blain-Nelson, P.; et al. Sex-related differences in age-associated downregulation of human ventricular myocardial β1-adrenergic receptors. J. Heart Lung Transplant. 2016, 35, 352–361. [Google Scholar] [CrossRef]
- López, M.G.; Abad, F.; Sancho, C.; de Pascual, R.; Borges, R.; Maroto, R.; Dixon, W.; Garcia, A.G. Membrane-mediated effects of the steroid 17-alpha-estradiol on adrenal catecholamine release. J. Pharmacol. Exp. Ther. 1991, 259, 279–285. [Google Scholar]
- Park, Y.H.; Cho, G.S.; Cho, E.T.; Park, Y.K.; Lee, M.J.; Chung, J.Y.; Hong, S.P.; Lee, J.J.; Jang, Y.; Yoo, H.J.; et al. Influence of 17- alpha-estradiol on catecholamine secretion from the perfused rat adrenal gland. Korean J. Intern. Med. 1996, 11, 25–39. [Google Scholar] [CrossRef]
- Gomes, H.L.; Graceli, J.B.; Gonçalves, W.L.; dos Santos, R.L.; Abreu, G.R.; Bissoli, N.S.; Pires, J.G.; Cicilini, M.A.; Moysés, M.R. Influence of gender and estrous cycle on plasma and renal catecholamine levels in rats. Can. J. Physiol. Pharmacol. 2012, 90, 75–82. [Google Scholar] [CrossRef]
- Matarrese, P.; Maccari, S.; Vona, R.; Gambardella, L.; Stati, T.; Marano, G. Role of β-Adrenergic Receptors and Estrogen in Cardiac Repair after Myocardial Infarction: An Overview. Int. J. Mol. Sci. 2021, 22, 8957. [Google Scholar] [CrossRef] [PubMed]
- Machuki, J.O.; Zhang, H.Y.; Harding, S.E.; Sun, H. Molecular pathways of oestrogen receptors and β-adrenergic receptors in cardiac cells: Recognition of their similarities, interactions and therapeutic value. Acta Physiol. 2018, 222, e12978. [Google Scholar] [CrossRef] [PubMed]
- Arosio, B.; Corbi, G.; Davinelli, S.; Giordano, V.; Liccardo, D.; Rapacciuolo, A.; Cannavo, A. Sex Differences in Cardiovascular Diseases: A Matter of Estrogens, Ceramides, and Sphingosine 1-Phosphate. Int. J. Mol. Sci. 2022, 23, 4009. [Google Scholar] [CrossRef] [PubMed]
- Blum, I.; Vered, Y.; Lifshitz, A.; Harel, D.; Blum, M.; Nordenberg, Y.; Harsat, A.; Sulkes, J.; Gabbay, U.; Graff, E. The effect of estrogen replacement therapy on plasma serotonin and catecholamines of postmenopausal women. Isr J. Med. Sci. 1996, 32, 1158–1162. [Google Scholar] [PubMed]
- Ferrer, M.; Meyer, M.; Osol, G. Estrogen replacement increases beta-adrenoceptor-mediated relaxation of rat mesenteric arteries. J. Vasc. Res. 1996, 33, 124–131. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Manson, J.E.; Kaunitz, A.M.; Anderson, G.L. Lessons learned from the Women’s Health Initiative trials of menopausal hormone therapy. Obstet. Gynecol. 2013, 121, 172–176. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Moreira, A.C.; Silva, A.M.; Santos, M.S.; Sardão, V.A. Phytoestrogens as alternative hormone replacement therapy in menopause: What is real, what is unknown. J. Steroid Biochem. Mol. Biol. 2014, 143, 61–71. [Google Scholar] [CrossRef]
- Franco, O.H.; Chowdhury, R.; Troup, J.; Voortman, T.; Kunutsor, S.; Kavousi, M.; Oliver-Williams, C.; Muka, T. Use of Plant-Based Therapies and Menopausal Symptoms: A Systematic Review and Meta-analysis. JAMA 2016, 315, 2554–2563. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.C.; Na, G.M.; Lim, D.Y. Resveratrol inhibits nicotinic stimulation-evoked catecholamine release from the adrenal medulla. Korean J. Physiol. Pharmacol. 2008, 12, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, Y.; Toyohira, Y.; Ueno, S.; Liu, M.; Tsutsui, M.; Yanagihara, N. Effects of Resveratrol, a grape polyphenol, on catecholamine secretion and synthesis in cultured bovine adrenal medullary cells. Biochem. Pharmacol. 2007, 74, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Morales, J.C.; Yáñez, M.; Orallo, F.; Cortés, L.; González, J.C.; Hernández-Guijo, J.M.; García, A.G.; de Diego, A.M. Blockade by nanomolar resveratrol of quantal catecholamine release in chromaffin cells. Mol. Pharmacol. 2010, 78, 734–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, B.; Maguy, A.; Clément, R.; Gosselin, H.; Poulin, F.; Ethier, N.; Tardif, J.C.; Hébert, T.E.; Calderone, A.; Nattel, S. Effects of resveratrol (trans-3,5,4′-trihydroxystilbene) treatment on cardiac remodeling following myocardial infarction. J Pharmacol. Exp. Ther. 2007, 323, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Benovic, J.L. G-protein-coupled receptors signal victory. Cell 2012, 151, 1148–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaillon, P. Relevance of intrinsic sympathomimetic activity for beta blockers. Am. J. Cardiol. 1990, 66, 21C–23C. [Google Scholar] [CrossRef]
- Bristow, M.R.; Larrabee, P.; Minobe, W.; Roden, R.; Skerl, L.; Klein, J.; Handwerger, D.; Port, J.D.; Müller-Beckmann, B. Receptor pharmacology of carvedilol in the human heart. J. Cardiovasc. Pharmacol. 1992, 19, S68–S80. [Google Scholar] [CrossRef]
- Chidiac, P.; Hebert, T.E.; Valiquette, M.; Dennis, M.; Bouvier, M. Inverse agonist activity of beta-adrenergic antagonists. Mol. Pharmacol. 1994, 45, 490–499. [Google Scholar]
- Bugiardini, R.; Yoon, J.; Kedev, S.; Stankovic, G.; Vasiljevic, Z.; Miličić, D.; Manfrini, O.; van der Schaar, M.; Gale, C.P.; Badimon, L.; et al. Prior Beta-Blocker Therapy for Hypertension and Sex-Based Differences in Heart Failure Among Patients with Incident Coronary Heart Disease. Hypertension 2020, 76, 819–826. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liccardo, D.; Arosio, B.; Corbi, G.; Cannavo, A. Sex/Gender- and Age-Related Differences in β-Adrenergic Receptor Signaling in Cardiovascular Diseases. J. Clin. Med. 2022, 11, 4280. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11154280
Liccardo D, Arosio B, Corbi G, Cannavo A. Sex/Gender- and Age-Related Differences in β-Adrenergic Receptor Signaling in Cardiovascular Diseases. Journal of Clinical Medicine. 2022; 11(15):4280. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11154280
Chicago/Turabian StyleLiccardo, Daniela, Beatrice Arosio, Graziamaria Corbi, and Alessandro Cannavo. 2022. "Sex/Gender- and Age-Related Differences in β-Adrenergic Receptor Signaling in Cardiovascular Diseases" Journal of Clinical Medicine 11, no. 15: 4280. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11154280