
Meeting the Challenges of Myocarditis: New Opportunities for Prevention, Detection, and Intervention—A Report from the 2021 National Heart, Lung, and Blood Institute Workshop

 , , ,
on behalf of the Workshop Speakers
, , ,
on behalf of the Workshop Speakers
Abstract
:1. Introduction
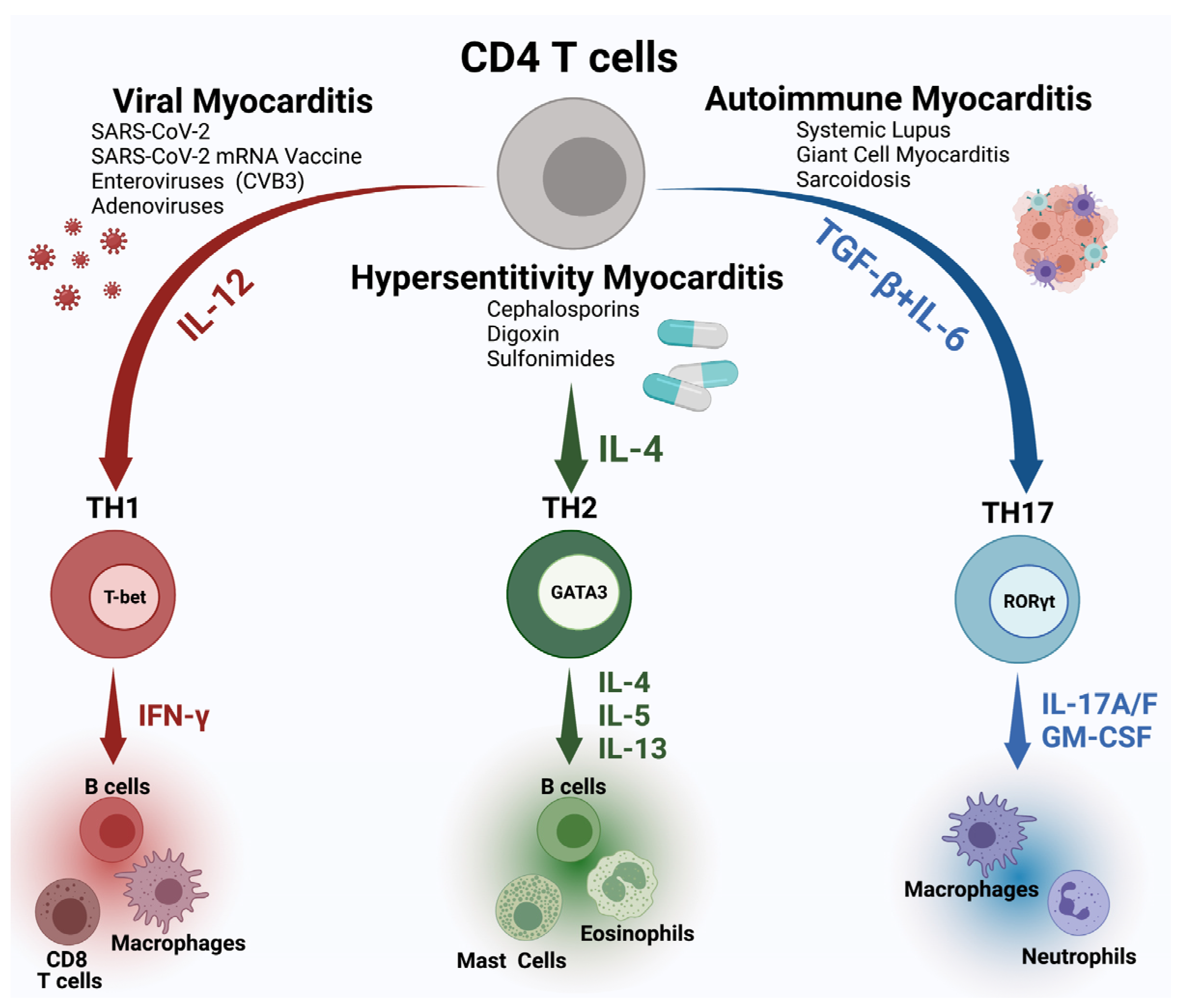
2. Etiologies of Myocarditis
- Although viruses are the most common cause of myocarditis, RT-PCR and genomic sequencing testing for viruses in biopsies may not correlate with active infection.
- Therefore, greater understanding is needed of the mechanisms of specific viruses for myocarditis pathogenesis. Specifically, research is needed to understand the factors which determine when acute viral myocarditis progresses to an autoimmune cardiomyopathy in humans.
- Understanding the mechanism of mRNA vaccine-induced myocarditis is important for the use of this technology to prevent COVID-19 and to understand the risks of other vaccines based on the same technology.
- It is critical to understand the broader scope of pathogenesis of inflammatory myocardial diseases in general is critical.
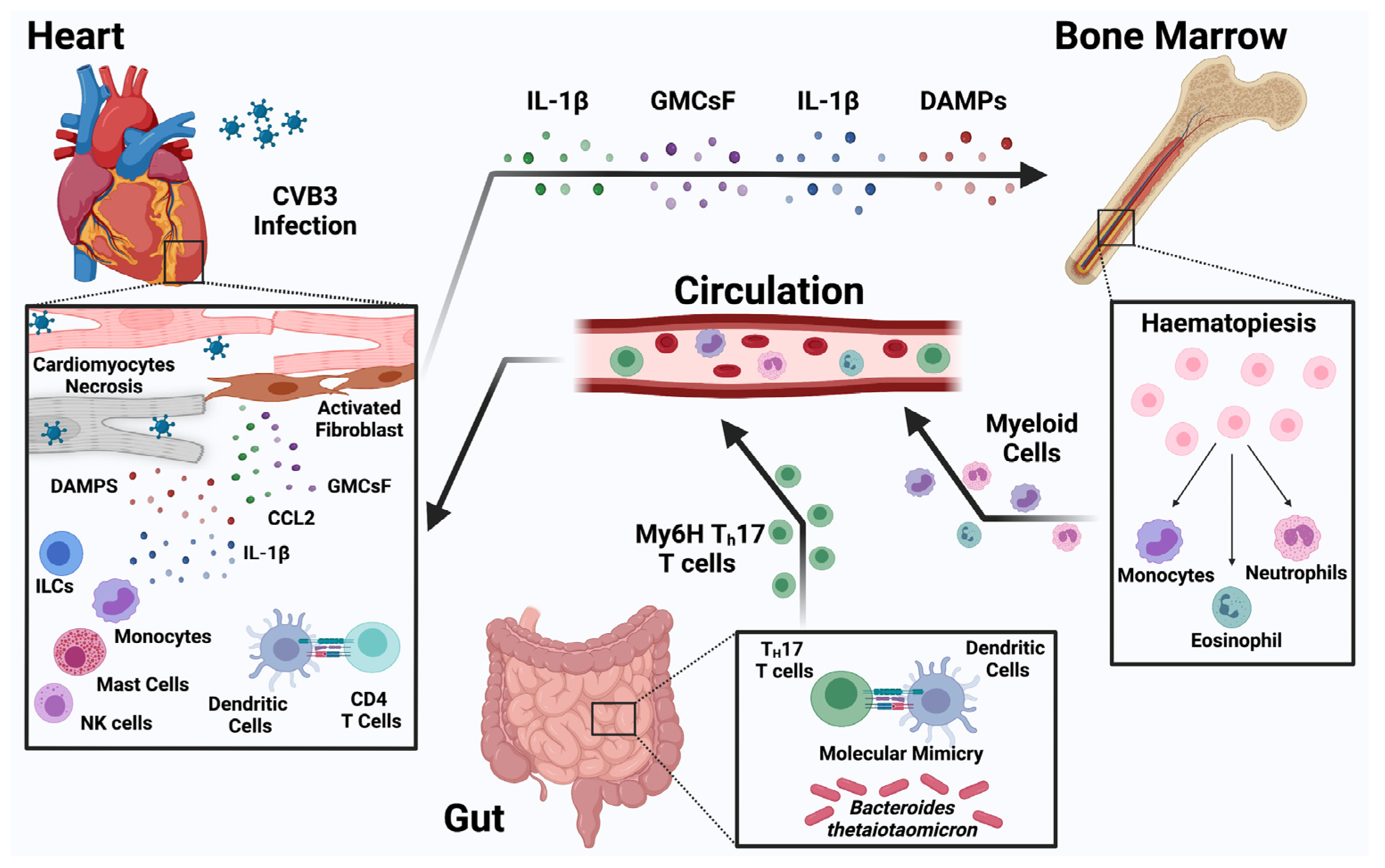
3. Pathogenesis of Myocarditis
- Further research into the role of innate and adaptive immune response in myocarditis and cardiac injury in both viral and autoimmune myocarditis is essential for the discovery of new diagnostics and treatments for myocarditis.
- Cardiac injury during the COVID-19 pandemic has revealed gaps in the knowledge of cardiac inflammation including the heterogeneity of cellular signaling, temporal sequence and regulation of inflammatory processes in cardiac tissues. New diagnostic methods including single cell sequencing linked to deep clinical phenotyping are needed to advance out understanding and identify new and potentially druggable targets.
- Lack of systemic and heart-specific immunophenotyping tools that can be deployed at the bedside are needed to understand distinct mechanism-based subgroups and develop more specific treatment strategies with acceptable risks.
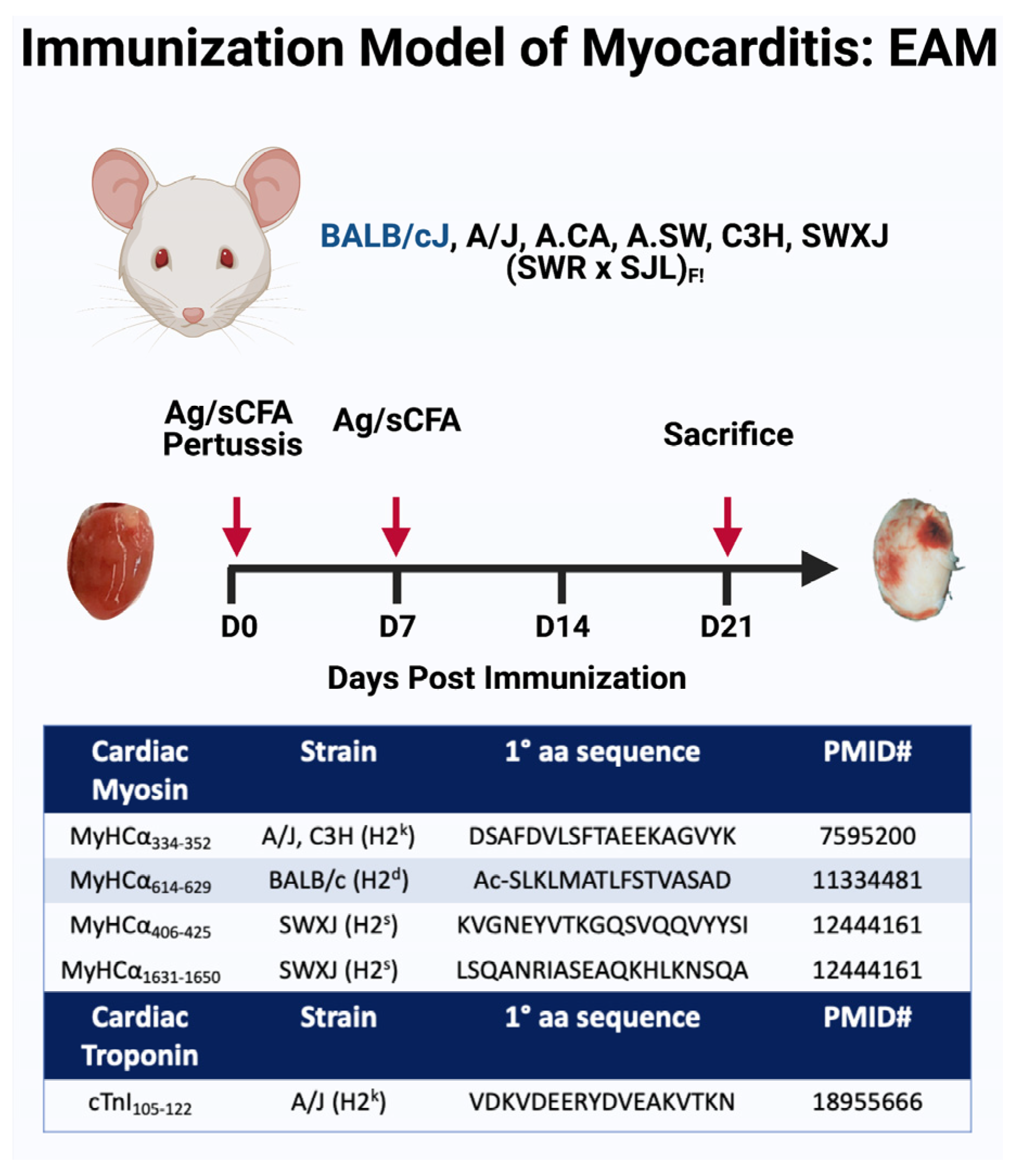
4. Animal Models
- A broader spectrum of myocarditis models of different etiologies are needed to understand emerging causes of myocarditis including COVID-19 myocarditis, ICI-induced myocarditis, and an mRNA vaccine-induced myocarditis.
- Efforts to standardize myocarditis animal models across different labs would improve the translatability of the basic research to bedside medicine.
- Higher utilization of an in vitro approach and organelles development would aid the study of some aspects of myocarditis pathogenesis.
5. Genetic Regulation of Myocarditis
- The role of non-immune genes in susceptibility to all types of myocarditis including viral myocarditis should be further studied.
- It is essential to examine the clinical consequences for the susceptibility to myocarditis and differences in immune response using in vivo and in vitro models.
- Prospective multi-center studies on the genetic role in myocarditis susceptibility are needed to capture the impact on disease severity and long terms outcomes.
- The establishment of an international myocarditis registry to collect genetic information and link it to patients’ clinical phenotypes and outcomes might be an important and initial step toward advancing research in genetic regulation of myocarditis.
6. Clinical Presentation
- A specific and sensitive mechanism-based diagnostic criteria that refines the detection of myocarditis across the spectrum of clinical presentations is a high priority gap.
- Efforts to quantify the impact of SDoH (e.g., using PhenX Toolkit or other survey or screening tools) on cardiovascular outcomes in myocarditis outcomes are necessary.
- Validation of tools to capture patient reported outcomes and quantitate psychosocial aspects of heath are needed to assess disease burden in diverse populations.
7. Diagnostic Imaging for Myocarditis
- The nonspecific presentation of myocarditis mandates improved discriminatory diagnostic testing using molecular targets for specific etiologies.
- Molecular targets of inflammation should be leveraged to improve the specificity of imaging methods.
- Integration of clinical imaging and immunological data within AI-assisted risk prediction models should be explored to improve risk assessment models.
8. New Diagnostic and Therapeutic Targets for Myocarditis
- Proteomic studies on archived samples of patients with myocarditis should be integrated with genetic data and cardiac imaging to deep phenotype patients with myocarditis and guide the development of new diagnostic and therapeutic targets.
- An improved understanding through model systems and patients at risk for developing of ICI-associated myocarditis are needed to dissect the pathogenesis of ICI myocarditis and design the next generation of treatment trials.
9. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclosure Statement
Disclaimer
References
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar] [CrossRef] [PubMed]
- National Heart, L. Blood Institute (NHLBI). Meeting the Challenges of Myocarditis: New Opportunities for Prevention, Detection, and Intervention Workshop- Executive Summary. Available online: https://www.nhlbi.nih.gov/events/2021/meeting-challenges-myocarditis-workshop (accessed on 6 January 2022).
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Tschope, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hubner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Verdonschot, J.; Hazebroek, M.; Merken, J.; Debing, Y.; Dennert, R.; Brunner-La Rocca, H.P.; Heymans, S. Relevance of cardiac parvovirus B19 in myocarditis and dilated cardiomyopathy: Review of the literature. Eur J. Heart Fail. 2016, 18, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.; Zidar, D.A.; Bristow, M.R.; Cameron, S.J.; Chan, T.; Harding, C.V., 3rd; Kwon, D.H.; Singh, T.; Tilton, J.C.; Tsai, E.J.; et al. COVID-19 and Cardiovascular Disease: From Bench to Bedside. Circ. Res. 2021, 128, 1214–1236. [Google Scholar] [CrossRef] [PubMed]
- Bearse, M.; Hung, Y.P.; Krauson, A.J.; Bonanno, L.; Boyraz, B.; Harris, C.K.; Helland, T.L.; Hilburn, C.F.; Hutchison, B.; Jobbagy, S.; et al. Factors associated with myocardial SARS-CoV-2 infection, myocarditis, and cardiac inflammation in patients with COVID-19. Mod. Pathol. 2021, 34, 1345–1357. [Google Scholar] [CrossRef]
- Fox, S.E.; Falgout, L.; Vander Heide, R.S. COVID-19 myocarditis: Quantitative analysis of the inflammatory infiltrate and a proposed mechanism. Cardiovasc. Pathol. 2021, 54, 107361. [Google Scholar] [CrossRef]
- Halushka, M.K.; Vander Heide, R.S. Myocarditis is rare in COVID-19 autopsies: Cardiovascular findings across 277 postmortem examinations. Cardiovasc. Pathol. 2021, 50, 107300. [Google Scholar] [CrossRef]
- Pellegrini, D.; Kawakami, R.; Guagliumi, G.; Sakamoto, A.; Kawai, K.; Gianatti, A.; Nasr, A.; Kutys, R.; Guo, L.; Cornelissen, A.; et al. Microthrombi as a Major Cause of Cardiac Injury in COVID-19: A Pathologic Study. Circulation 2021, 143, 1031–1042. [Google Scholar] [CrossRef]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L.; et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef]
- Gurdogan, M.; Yalta, K. Myocarditis associated with immune checkpoint inhibitors: Practical considerations in diagnosis and management. Anatol. J. Cardiol. 2020, 24, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Halsell, J.S.; Riddle, J.R.; Atwood, J.E.; Gardner, P.; Shope, R.; Poland, G.A.; Gray, G.C.; Ostroff, S.; Eckart, R.E.; Hospenthal, D.R.; et al. Myopericarditis following smallpox vaccination among vaccinia-naive US military personnel. JAMA 2003, 289, 3283–3289. [Google Scholar] [CrossRef] [PubMed]
- Casey, C.G.; Iskander, J.K.; Roper, M.H.; Mast, E.E.; Wen, X.J.; Torok, T.J.; Chapman, L.E.; Swerdlow, D.L.; Morgan, J.; Heffelfinger, J.D.; et al. Adverse events associated with smallpox vaccination in the United States, January-October 2003. JAMA 2005, 294, 2734–2743. [Google Scholar] [CrossRef]
- Su, J.R.; McNeil, M.M.; Welsh, K.J.; Marquez, P.L.; Ng, C.; Yan, M.; Cano, M.V. Myopericarditis after vaccination, Vaccine Adverse Event Reporting System (VAERS), 1990–2018. Vaccine 2021, 39, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, J.; Crane, B.; Weinmann, S.; Naleway, A.L.; Vaccine Safety Datalink Investigator Team. Myocarditis and pericarditis are rare following live viral vaccinations in adults. Vaccine 2018, 36, 1524–1527. [Google Scholar] [CrossRef]
- Mevorach, D.; Anis, E.; Cedar, N.; Bromberg, M.; Haas, E.J.; Nadir, E.; Olsha-Castell, S.; Arad, D.; Hasin, T.; Levi, N.; et al. Myocarditis after BNT162b2 mRNA Vaccine against Covid-19 in Israel. N. Engl. J. Med. 2021, 385, 2140–2149. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.; Ryan, M.; Engler, R.; Hoffman, D.; McClenathan, B.; Collins, L.; Loran, D.; Hrncir, D.; Herring, K.; Platzer, M.; et al. Myocarditis Following Immunization With mRNA COVID-19 Vaccines in Members of the US Military. JAMA Cardiol. 2021, 6, 1202–1206. [Google Scholar] [CrossRef]
- Witberg, G.; Barda, N.; Hoss, S.; Richter, I.; Wiessman, M.; Aviv, Y.; Grinberg, T.; Auster, O.; Dagan, N.; Balicer, R.D.; et al. Myocarditis after Covid-19 Vaccination in a Large Health Care Organization. N. Engl. J. Med. 2021, 385, 2132–2139. [Google Scholar] [CrossRef]
- Verma, A.K.; Lavine, K.J.; Lin, C.Y. Myocarditis after Covid-19 mRNA Vaccination. N. Engl. J. Med. 2021, 385, 1332–1334. [Google Scholar] [CrossRef]
- Cooper, L.T., Jr.; Berry, G.J.; Shabetai, R. Idiopathic giant-cell myocarditis--natural history and treatment. Multicenter Giant Cell Myocarditis Study Group Investigators. N. Engl. J. Med. 1997, 336, 1860–1866. [Google Scholar] [CrossRef]
- Cooper, L.T., Jr.; Berry, G.J.; Rizeq, M.; Schroeder, J.S. Giant cell myocarditis. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 1995, 14, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; Madden, B.; Spyrou, N.; Pomerance, A.; Mitchell, A.; Yacoub, M. Response of recurrent giant cell myocarditis in a transplanted heart to intensive immunosuppression. Eur. Heart J. 1991, 12, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Gries, W.; Farkas, D.; Winters, G.L.; Costanzo-Nordin, M.R. Giant cell myocarditis: First report of disease recurrence in the transplanted heart. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 1992, 11, 370–374. [Google Scholar]
- Grant, S.C. Giant cell myocarditis in a transplanted heart. Eur. Heart J. 1993, 14, 1437. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.C. Recurrent giant cell myocarditis after transplantation. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 1993, 12, 155–156. [Google Scholar]
- Busteed, S.; Sparrow, P.; Molloy, C.; Molloy, M.G. Myocarditis as a prognostic indicator in systemic lupus erythematosus. Postgrad. Med. J. 2004, 80, 366–367. [Google Scholar] [CrossRef]
- Levin, M.D.; Zoet-Nugteren, S.K.; Markusse, H.M. Myocarditis and primary Sjogren's syndrome. Lancet 1999, 354, 128–129. [Google Scholar] [CrossRef]
- Gilotra, N.; Okada, D.; Sharma, A.; Chrispin, J. Management of Cardiac Sarcoidosis in 2020. Arrhythm. Electrophysiol. Rev. 2020, 9, 182–188. [Google Scholar] [CrossRef]
- Cunningham, M.W.; Antone, S.M.; Gulizia, J.M.; McManus, B.M.; Fischetti, V.A.; Gauntt, C.J. Cytotoxic and viral neutralizing antibodies crossreact with streptococcal M protein, enteroviruses, and human cardiac myosin. Proc. Natl. Acad. Sci. USA 1992, 89, 1320–1324. [Google Scholar] [CrossRef]
- Huber, S.A.; Cunningham, M.W. Streptococcal M protein peptide with similarity to myosin induces CD4+ T cell-dependent myocarditis in MRL/++ mice and induces partial tolerance against coxsakieviral myocarditis. J. Immunol. 1996, 156, 3528–3534. [Google Scholar]
- Bracamonte-Baran, W.; Cihakova, D. Cardiac Autoimmunity: Myocarditis. Adv. Exp. Med. Biol. 2017, 1003, 187–221. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Chen, G.; Bracamonte-Baran, W.; Choi, H.S.; Diny, N.L.; Sung, J.; Hughes, D.; Won, T.; Wood, M.K.; Talor, M.V.; et al. The Cardiac Microenvironment Instructs Divergent Monocyte Fates and Functions in Myocarditis. Cell Rep. 2019, 28, 172–189.e17. [Google Scholar] [CrossRef] [PubMed]
- Baldeviano, G.C.; Barin, J.G.; Talor, M.V.; Srinivasan, S.; Bedja, D.; Zheng, D.; Gabrielson, K.; Iwakura, Y.; Rose, N.R.; Cihakova, D. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ. Res. 2010, 106, 1646–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Ong, S.; Talor, M.V.; Barin, J.G.; Baldeviano, G.C.; Kass, D.A.; Bedja, D.; Zhang, H.; Sheikh, A.; Margolick, J.B.; et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J. Exp. Med. 2014, 211, 1449–1464. [Google Scholar] [CrossRef]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef]
- Clemente-Casares, X.; Hosseinzadeh, S.; Barbu, I.; Dick, S.A.; Macklin, J.A.; Wang, Y.; Momen, A.; Kantores, C.; Aronoff, L.; Farno, M.; et al. A CD103(+) Conventional Dendritic Cell Surveillance System Prevents Development of Overt Heart Failure during Subclinical Viral Myocarditis. Immunity 2017, 47, 974–989.e8. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Gil-Cruz, C.; Perez-Shibayama, C.; De Martin, A.; Ronchi, F.; van der Borght, K.; Niederer, R.; Onder, L.; Lutge, M.; Novkovic, M.; Nindl, V.; et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science 2019, 366, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Afanasyeva, M.; Wang, Y.; Kaya, Z.; Stafford, E.A.; Dohmen, K.M.; Sadighi Akha, A.A.; Rose, N.R. Interleukin-12 receptor/STAT4 signaling is required for the development of autoimmune myocarditis in mice by an interferon-gamma-independent pathway. Circulation 2001, 104, 3145–3151. [Google Scholar] [CrossRef] [PubMed]
- Sonderegger, I.; Rohn, T.A.; Kurrer, M.O.; Iezzi, G.; Zou, Y.; Kastelein, R.A.; Bachmann, M.F.; Kopf, M. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur. J. Immunol. 2006, 36, 2849–2856. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Hafler, D.A. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin. Immunol. 2013, 25, 305–312. [Google Scholar] [CrossRef]
- Wu, L.; Diny, N.L.; Ong, S.; Barin, J.G.; Hou, X.; Rose, N.R.; Talor, M.V.; Cihakova, D. Pathogenic IL-23 signaling is required to initiate GM-CSF-driven autoimmune myocarditis in mice. Eur. J. Immunol. 2016, 46, 582–592. [Google Scholar] [CrossRef]
- Rangachari, M.; Mauermann, N.; Marty, R.R.; Dirnhofer, S.; Kurrer, M.O.; Komnenovic, V.; Penninger, J.M.; Eriksson, U. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J. Exp. Med. 2006, 203, 2009–2019. [Google Scholar] [CrossRef]
- Fairweather, D.; Frisancho-Kiss, S.; Yusung, S.A.; Barrett, M.A.; Davis, S.E.; Gatewood, S.J.; Njoku, D.B.; Rose, N.R. Interferon-gamma protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines transforming growth factor-beta 1, interleukin-1 beta, and interleukin-4 in the heart. Am. J. Pathol. 2004, 165, 1883–1894. [Google Scholar] [CrossRef]
- Cihakova, D.; Barin, J.G.; Afanasyeva, M.; Kimura, M.; Fairweather, D.; Berg, M.; Talor, M.V.; Baldeviano, G.C.; Frisancho, S.; Gabrielson, K.; et al. Interleukin-13 protects against experimental autoimmune myocarditis by regulating macrophage differentiation. Am. J. Pathol. 2008, 172, 1195–1208. [Google Scholar] [CrossRef]
- Afanasyeva, M.; Wang, Y.; Kaya, Z.; Park, S.; Zilliox, M.J.; Schofield, B.H.; Hill, S.L.; Rose, N.R. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am. J. Pathol. 2001, 159, 193–203. [Google Scholar] [CrossRef]
- Chen, P.; Baldeviano, G.C.; Ligons, D.L.; Talor, M.V.; Barin, J.G.; Rose, N.R.; Cihakova, D. Susceptibility to autoimmune myocarditis is associated with intrinsic differences in CD4(+) T cells. Clin. Exp. Immunol. 2012, 169, 79–88. [Google Scholar] [CrossRef]
- Myers, J.M.; Cooper, L.T.; Kem, D.C.; Stavrakis, S.; Kosanke, S.D.; Shevach, E.M.; Fairweather, D.; Stoner, J.A.; Cox, C.J.; Cunningham, M.W. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight 2016, 1, e85851. [Google Scholar] [CrossRef] [PubMed]
- Diny, N.L.; Baldeviano, G.C.; Talor, M.V.; Barin, J.G.; Ong, S.; Bedja, D.; Hays, A.G.; Gilotra, N.A.; Coppens, I.; Rose, N.R.; et al. Eosinophil-derived IL-4 drives progression of myocarditis to inflammatory dilated cardiomyopathy. J. Exp. Med. 2017, 214, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Diny, N.L.; Hou, X.; Barin, J.G.; Chen, G.; Talor, M.V.; Schaub, J.; Russell, S.D.; Klingel, K.; Rose, N.R.; Cihakova, D. Macrophages and cardiac fibroblasts are the main producers of eotaxins and regulate eosinophil trafficking to the heart. Eur. J. Immunol. 2016, 46, 2749–2760. [Google Scholar] [CrossRef] [Green Version]
- Adamo, L.; Rocha-Resende, C.; Lin, C.Y.; Evans, S.; Williams, J.; Dun, H.; Li, W.; Mpoy, C.; Andhey, P.S.; Rogers, B.E.; et al. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight 2020, 5, e134700. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Helio, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef] [PubMed]
- Herda, L.R.; Trimpert, C.; Nauke, U.; Landsberger, M.; Hummel, A.; Beug, D.; Kieback, A.; Dorr, M.; Empen, K.; Knebel, F.; et al. Effects of immunoadsorption and subsequent immunoglobulin G substitution on cardiopulmonary exercise capacity in patients with dilated cardiomyopathy. Am. Heart J. 2010, 159, 809–816. [Google Scholar] [CrossRef]
- Mascaro-Blanco, A.; Alvarez, K.; Yu, X.; Lindenfeld, J.; Olansky, L.; Lyons, T.; Duvall, D.; Heuser, J.S.; Gosmanova, A.; Rubenstein, C.J.; et al. Consequences of unlocking the cardiac myosin molecule in human myocarditis and cardiomyopathies. Autoimmunity 2008, 41, 442–453. [Google Scholar] [CrossRef]
- Abston, E.D.; Barin, J.G.; Cihakova, D.; Bucek, A.; Coronado, M.J.; Brandt, J.E.; Bedja, D.; Kim, J.B.; Georgakopoulos, D.; Gabrielson, K.L.; et al. IL-33 independently induces eosinophilic pericarditis and cardiac dilation: ST2 improves cardiac function. Circ. Heart Fail. 2012, 5, 366–375. [Google Scholar] [CrossRef]
- Choi, H.S.; Won, T.; Hou, X.; Chen, G.; Bracamonte-Baran, W.; Talor, M.V.; Jurcova, I.; Szarszoi, O.; Curnova, L.; Striz, I.; et al. Innate Lymphoid Cells Play a Pathogenic Role in Pericarditis. Cell Rep. 2020, 30, 2989–3003.e6. [Google Scholar] [CrossRef]
- Coronado, M.J.; Bruno, K.A.; Blauwet, L.A.; Tschope, C.; Cunningham, M.W.; Pankuweit, S.; van Linthout, S.; Jeon, E.S.; McNamara, D.M.; Krejci, J.; et al. Elevated Sera sST2 Is Associated With Heart Failure in Men </= 50 Years Old With Myocarditis. J. Am. Heart Assoc. 2019, 8, e008968. [Google Scholar] [CrossRef]
- Fairweather, D.; Cooper, L.T., Jr.; Blauwet, L.A. Sex and gender differences in myocarditis and dilated cardiomyopathy. Curr. Probl. Cardiol. 2013, 38, 7–46. [Google Scholar] [CrossRef]
- Oertelt-Prigione, S. The influence of sex and gender on the immune response. Autoimmun. Rev. 2012, 11, A479–A485. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.; Yusung, S.; Frisancho, S.; Barrett, M.; Gatewood, S.; Steele, R.; Rose, N.R. IL-12 receptor beta 1 and Toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J. Immunol. 2003, 170, 4731–4737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisancho-Kiss, S.; Davis, S.E.; Nyland, J.F.; Frisancho, J.A.; Cihakova, D.; Barrett, M.A.; Rose, N.R.; Fairweather, D. Cutting edge: Cross-regulation by TLR4 and T cell Ig mucin-3 determines sex differences in inflammatory heart disease. J. Immunol. 2007, 178, 6710–6714. [Google Scholar] [CrossRef] [PubMed]
- Lyden, D.C.; Olszewski, J.; Feran, M.; Job, L.P.; Huber, S.A. Coxsackievirus B-3-induced myocarditis. Effect of sex steroids on viremia and infectivity of cardiocytes. Am. J. Pathol. 1987, 126, 432–438. [Google Scholar]
- Roberts, B.J.; Moussawi, M.; Huber, S.A. Sex differences in TLR2 and TLR4 expression and their effect on coxsackievirus-induced autoimmune myocarditis. Exp. Mol. Pathol. 2013, 94, 58–64. [Google Scholar] [CrossRef]
- Blanco-Dominguez, R.; Sanchez-Diaz, R.; de la Fuente, H.; Jimenez-Borreguero, L.J.; Matesanz-Marin, A.; Relano, M.; Jimenez-Alejandre, R.; Linillos-Pradillo, B.; Tsilingiri, K.; Martin-Mariscal, M.L.; et al. A Novel Circulating MicroRNA for the Detection of Acute Myocarditis. N. Engl. J. Med. 2021, 384, 2014–2027. [Google Scholar] [CrossRef]
- Omur, O.; Erdogan, M.; Ozkilic, H.; Yilmaz, C. Scintigraphic methods to evaluate alterations of gastric and esophageal functions in female obesity. Mol. Imaging Radionucl. Ther. 2014, 23, 5–11. [Google Scholar] [CrossRef]
- Coronado, M.J.; Brandt, J.E.; Kim, E.; Bucek, A.; Bedja, D.; Abston, E.D.; Shin, J.; Gabrielson, K.L.; Mitzner, W.; Fairweather, D. Testosterone and interleukin-1beta increase cardiac remodeling during coxsackievirus B3 myocarditis via serpin A 3n. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1726–H1736. [Google Scholar] [CrossRef]
- Huber, S.A.; Kupperman, J.; Newell, M.K. Estradiol prevents and testosterone promotes Fas-dependent apoptosis in CD4+ Th2 cells by altering Bcl 2 expression. Lupus 1999, 8, 384–387. [Google Scholar] [CrossRef]
- Huber, S.A.; Kupperman, J.; Newell, M.K. Hormonal regulation of CD4(+) T-cell responses in coxsackievirus B3-induced myocarditis in mice. J. Virol. 1999, 73, 4689–4695. [Google Scholar] [CrossRef]
- Fuse, K.; Chan, G.; Liu, Y.; Gudgeon, P.; Husain, M.; Chen, M.; Yeh, W.C.; Akira, S.; Liu, P.P. Myeloid differentiation factor-88 plays a crucial role in the pathogenesis of Coxsackievirus B3-induced myocarditis and influences type I interferon production. Circulation 2005, 112, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.A.; Gauntt, C.J.; Sakkinen, P. Enteroviruses and myocarditis: Viral pathogenesis through replication, cytokine induction, and immunopathogenicity. Adv. Virus Res. 1998, 51, 35–80. [Google Scholar] [PubMed]
- Li, Y.; Heuser, J.S.; Kosanke, S.D.; Hemric, M.; Cunningham, M.W. Cryptic epitope identified in rat and human cardiac myosin S2 region induces myocarditis in the Lewis rat. J. Immunol. 2004, 172, 3225–3234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodama, M.; Matsumoto, Y.; Fujiwara, M.; Masani, F.; Izumi, T.; Shibata, A. A novel experimental model of giant cell myocarditis induced in rats by immunization with cardiac myosin fraction. Clin. Immunol. Immunopathol. 1990, 57, 250–262. [Google Scholar] [CrossRef]
- Anzai, A.; Mindur, J.E.; Halle, L.; Sano, S.; Choi, J.L.; He, S.; McAlpine, C.S.; Chan, C.T.; Kahles, F.; Valet, C.; et al. Self-reactive CD4(+) IL-3(+) T cells amplify autoimmune inflammation in myocarditis by inciting monocyte chemotaxis. J. Exp. Med. 2019, 216, 369–383. [Google Scholar] [CrossRef]
- Cihakova, D.; Sharma, R.B.; Fairweather, D.; Afanasyeva, M.; Rose, N.R. Animal models for autoimmune myocarditis and autoimmune thyroiditis. Methods Mol. Med. 2004, 102, 175–193. [Google Scholar] [CrossRef]
- Goser, S.; Andrassy, M.; Buss, S.J.; Leuschner, F.; Volz, C.H.; Ottl, R.; Zittrich, S.; Blaudeck, N.; Hardt, S.E.; Pfitzer, G.; et al. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation 2006, 114, 1693–1702. [Google Scholar] [CrossRef]
- Barin, J.G.; Baldeviano, G.C.; Talor, M.V.; Wu, L.; Ong, S.; Fairweather, D.; Bedja, D.; Stickel, N.R.; Fontes, J.A.; Cardamone, A.B.; et al. Fatal eosinophilic myocarditis develops in the absence of IFN-gamma and IL-17A. J. Immunol. 2013, 191, 4038–4047. [Google Scholar] [CrossRef]
- Prows, D.R.; Klingler, A.; Gibbons, W.J., Jr.; Homan, S.M.; Zimmermann, N. Characterization of a mouse model of hypereosinophilia-associated heart disease. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H405–H414. [Google Scholar] [CrossRef]
- Wang, J.; Okazaki, I.M.; Yoshida, T.; Chikuma, S.; Kato, Y.; Nakaki, F.; Hiai, H.; Honjo, T.; Okazaki, T. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 2010, 22, 443–452. [Google Scholar] [CrossRef]
- Wei, S.C.; Meijers, W.C.; Axelrod, M.L.; Anang, N.A.S.; Screever, E.M.; Wescott, E.C.; Johnson, D.B.; Whitley, E.; Lehmann, L.; Courand, P.Y.; et al. A Genetic Mouse Model Recapitulates Immune Checkpoint Inhibitor-Associated Myocarditis and Supports a Mechanism-Based Therapeutic Intervention. Cancer Discov. 2021, 11, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Carlquist, J.F.; Menlove, R.L.; Murray, M.B.; O’Connell, J.B.; Anderson, J.L. HLA class II (DR and DQ) antigen associations in idiopathic dilated cardiomyopathy. Validation study and meta-analysis of published HLA association studies. Circulation 1991, 83, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Martinetti, M.; Dugoujon, J.M.; Caforio, A.L.; Schwarz, G.; Gavazzi, A.; Graziano, G.; Arbustini, E.; Lorini, R.; McKenna, W.J.; Bottazzo, G.F.; et al. HLA and immunoglobulin polymorphisms in idiopathic dilated cardiomyopathy. Human Immunol. 1992, 35, 193–199. [Google Scholar] [CrossRef]
- Li, H.S.; Ligons, D.L.; Rose, N.R. Genetic complexity of autoimmune myocarditis. Autoimmun. Rev. 2008, 7, 168–173. [Google Scholar] [CrossRef]
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.; Varela, J.; David, R.M.; Banks, L.; Huang, C.H.; Li, H.; et al. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223. [Google Scholar] [CrossRef]
- Wellington, D.; Laurenson-Schafer, H.; Abdel-Haq, A.; Dong, T. IFITM3: How genetics influence influenza infection demographically. Biomed. J. 2019, 42, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Noutsias, M.; Fechner, H.; de Jonge, H.; Wang, X.; Dekkers, D.; Houtsmuller, A.B.; Pauschinger, M.; Bergelson, J.; Warraich, R.; Yacoub, M.; et al. Human coxsackie-adenovirus receptor is colocalized with integrins alpha(v)beta(3) and alpha(v)beta(5) on the cardiomyocyte sarcolemma and upregulated in dilated cardiomyopathy: Implications for cardiotropic viral infections. Circulation 2001, 104, 275–280. [Google Scholar] [CrossRef]
- Piriou, N.; Marteau, L.; Kyndt, F.; Serfaty, J.M.; Toquet, C.; Le Gloan, L.; Warin-Fresse, K.; Guijarro, D.; Le Tourneau, T.; Conan, E.; et al. Familial screening in case of acute myocarditis reveals inherited arrhythmogenic left ventricular cardiomyopathies. ESC Heart Fail. 2020, 7, 1520–1533. [Google Scholar] [CrossRef]
- Ader, F.; Surget, E.; Charron, P.; Redheuil, A.; Zouaghi, A.; Maltret, A.; Marijon, E.; Denjoy, I.; Hermida, A.; Fressart, V.; et al. Inherited Cardiomyopathies Revealed by Clinically Suspected Myocarditis: Highlights from Genetic Testing. Circ. Genom. Precis. Med. 2020, 13, e002744. [Google Scholar] [CrossRef]
- Kontorovich, A.R.; Patel, N.; Moscati, A.; Richter, F.; Peter, I.; Purevjav, E.; Selejan, S.R.; Kindermann, I.; Towbin, J.A.; Bohm, M.; et al. Myopathic Cardiac Genotypes Increase Risk for Myocarditis. JACC Basic Transl. Sci. 2021, 6, 584–592. [Google Scholar] [CrossRef]
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of dilated cardiomyopathy: Practical implications for heart failure management. Nat. Rev. Cardiol. 2020, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.T., Jr.; Cihakova, D. Do Genes Influence Susceptibility to Myocarditis? JACC Basic Transl. Sci. 2021, 6, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Lee, G.H.; Badorff, C.; Dorner, A.; Lee, S.; Wolf, P.; Knowlton, K.U. Dystrophin deficiency markedly increases enterovirus-induced cardiomyopathy: A genetic predisposition to viral heart disease. Nat. Med. 2002, 8, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Ammirati, E.; Frigerio, M.; Adler, E.D.; Basso, C.; Birnie, D.H.; Brambatti, M.; Friedrich, M.G.; Klingel, K.; Lehtonen, J.; Moslehi, J.J.; et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy: An Expert Consensus Document. Circ. Heart Fail. 2020, 13, e007405. [Google Scholar] [CrossRef]
- Baritussio, A.; Schiavo, A.; Basso, C.; Giordani, A.S.; Cheng, C.Y.; Pontara, E.; Cattini, M.G.; Bison, E.; Gallo, N.; De Gaspari, M.; et al. Predictors of relapse, death or heart transplantation in myocarditis before the introduction of immunosuppression: Negative prognostic impact of female gender, fulminant onset, lower ejection fraction and serum autoantibodies. Eur. J. Heart Fail. 2022, 24, 1033–1044. [Google Scholar] [CrossRef]
- Havranek, E.P.; Mujahid, M.S.; Barr, D.A.; Blair, I.V.; Cohen, M.S.; Cruz-Flores, S.; Davey-Smith, G.; Dennison-Himmelfarb, C.R.; Lauer, M.S.; Lockwood, D.W.; et al. Social Determinants of Risk and Outcomes for Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2015, 132, 873–898. [Google Scholar] [CrossRef]
- Baumer, Y.; Farmer, N.; Premeaux, T.A.; Wallen, G.R.; Powell-Wiley, T.M. Health Disparities in COVID-19: Addressing the Role of Social Determinants of Health in Immune System Dysfunction to Turn the Tide. Front. Public Health 2020, 8, 559312. [Google Scholar] [CrossRef]
- Park, J.J.; Park, J.B.; Park, J.H.; Cho, G.Y. Global Longitudinal Strain to Predict Mortality in Patients with Acute Heart Failure. J. Am. Coll. Cardiol. 2018, 71, 1947–1957. [Google Scholar] [CrossRef]
- Gorcsan, J., 3rd; Tanaka, H. Echocardiographic assessment of myocardial strain. J. Am. Coll. Cardiol. 2011, 58, 1401–1413. [Google Scholar] [CrossRef]
- Caspar, T.; Fichot, M.; Ohana, M.; El Ghannudi, S.; Morel, O.; Ohlmann, P. Late Detection of Left Ventricular Dysfunction Using Two-Dimensional and Three-Dimensional Speckle-Tracking Echocardiography in Patients with History of Nonsevere Acute Myocarditis. J. Am. Soc. Echocardiogr. 2017, 30, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Kagiyama, N.; Hasselberg, N.E.; Blauwet, L.A.; Briller, J.; Cooper, L.; Fett, J.D.; Hsich, E.; Wells, G.; McNamara, D.; et al. Global Left Ventricular Strain at Presentation Is Associated with Subsequent Recovery in Patients with Peripartum Cardiomyopathy. J. Am. Soc. Echocardiogr. 2019, 32, 1565–1573. [Google Scholar] [CrossRef]
- Moreira, H.T.; Volpe, G.J.; Marin-Neto, J.A.; Nwabuo, C.C.; Ambale-Venkatesh, B.; Gali, L.G.; Almeida-Filho, O.C.; Romano, M.M.D.; Pazin-Filho, A.; Maciel, B.C.; et al. Right Ventricular Systolic Dysfunction in Chagas Disease Defined by Speckle-Tracking Echocardiography: A Comparative Study with Cardiac Magnetic Resonance Imaging. J. Am. Soc. Echocardiogr. 2017, 30, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Buxton, D.B.; Antman, M.; Danthi, N.; Dilsizian, V.; Fayad, Z.A.; Garcia, M.J.; Jaff, M.R.; Klimas, M.; Libby, P.; Nahrendorf, M.; et al. Report of the National Heart, Lung, and Blood Institute working group on the translation of cardiovascular molecular imaging. Circulation 2011, 123, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Sabahat Bokhari, M.; Lin, J.C.; Julien, H.M. FDG-PET Is a Superior Tool in the Diagnosis and Management of Cardiac Sarcoidosis. Available online: https://www.acc.org/latest-in-cardiology/articles/2017/04/10/08/43/fdg-pet-is-a-superior-tool (accessed on 6 January 2022).
- Balogh, V.; MacAskill, M.G.; Hadoke, P.W.F.; Gray, G.A.; Tavares, A.A.S. Positron Emission Tomography Techniques to Measure Active Inflammation, Fibrosis and Angiogenesis: Potential for Non-invasive Imaging of Hypertensive Heart Failure. Front. Cardiovasc. Med. 2021, 8, 719031. [Google Scholar] [CrossRef]
- Karamitsos, T.D.; Arvanitaki, A.; Karvounis, H.; Neubauer, S.; Ferreira, V.M. Myocardial Tissue Characterization and Fibrosis by Imaging. JACC Cardiovasc. Imaging 2020, 13, 1221–1234. [Google Scholar] [CrossRef]
- Ferreira, V.M.; Schulz-Menger, J.; Holmvang, G.; Kramer, C.M.; Carbone, I.; Sechtem, U.; Kindermann, I.; Gutberlet, M.; Cooper, L.T.; Liu, P.; et al. Cardiovascular Magnetic Resonance in Nonischemic Myocardial Inflammation: Expert Recommendations. J. Am. Coll. Cardiol. 2018, 72, 3158–3176. [Google Scholar] [CrossRef] [PubMed]
- Greulich, S.; Seitz, A.; Muller, K.A.L.; Grun, S.; Ong, P.; Ebadi, N.; Kreisselmeier, K.P.; Seizer, P.; Bekeredjian, R.; Zwadlo, C.; et al. Predictors of Mortality in Patients With Biopsy-Proven Viral Myocarditis: 10-Year Outcome Data. J. Am. Heart Assoc. 2020, 9, e015351. [Google Scholar] [CrossRef]
- Grani, C.; Biere, L.; Eichhorn, C.; Kaneko, K.; Agarwal, V.; Aghayev, A.; Steigner, M.; Blankstein, R.; Jerosch-Herold, M.; Kwong, R.Y. Incremental value of extracellular volume assessment by cardiovascular magnetic resonance imaging in risk stratifying patients with suspected myocarditis. Int. J. Cardiovasc. Imaging 2019, 35, 1067–1078. [Google Scholar] [CrossRef]
- Abbas, H.; Broche, L.M.; Ezdoglian, A.; Li, D.; Yuecel, R.; James Ross, P.; Cheyne, L.; Wilson, H.M.; Lurie, D.J.; Dawson, D.K. Fast field-cycling magnetic resonance detection of intracellular ultra-small iron oxide particles in vitro: Proof-of-concept. J. Magn. Reson. 2020, 313, 106722. [Google Scholar] [CrossRef]
- Rau, C.D.; Lusis, A.J.; Wang, Y. Systems Genetics for Mechanistic Discovery in Heart Diseases. Circ. Res. 2020, 126, 1795–1815. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.Y.; Pandey, A.K.; Maron, B.A.; Loscalzo, J. Network medicine in Cardiovascular Research. Cardiovasc. Res. 2021, 117, 2186–2202. [Google Scholar] [CrossRef]
- Leopold, J.A.; Maron, B.A.; Loscalzo, J. The application of big data to cardiovascular disease: Paths to precision medicine. J. Clin. Investig. 2020, 130, 29–38. [Google Scholar] [CrossRef]
- Porritt, R.A.; Binek, A.; Paschold, L.; Noval Rivas, M.; Mc Ardle, A.; Yonker, L.M.; Alter, G.; Chandnani, H.K.; Lopez, M.; Fasano, A.; et al. The autoimmune signature of hyperinflammatory multisystem inflammatory syndrome in children. J. Clin. Investig. 2021, 131, e151520. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.D.; Mohan, J.; Schneider, C.; Bajpai, G.; Purevjav, E.; Canter, C.E.; Towbin, J.; Bredemeyer, A.; Lavine, K.J. Pediatric and adult dilated cardiomyopathy represent distinct pathological entities. JCI Insight 2017, 2, e94382. [Google Scholar] [CrossRef] [PubMed]
- Mamic, P.; Chaikijurajai, T.; Tang, W.H.W. Gut microbiome—A potential mediator of pathogenesis in heart failure and its comorbidities: State-of-the-art review. J. Mol. Cell Cardiol. 2021, 152, 105–117. [Google Scholar] [CrossRef]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wulfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martinez, L.; Sanchez-Diaz, M.; Diaz-Garcia, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Transthoracic Echocardiography | Positron Emission Tomography | Cardiovascular Magnetic Resonance | |
|---|---|---|---|
| Ventricular volumes, myocardial mass, systolic function | +++ | ++ | ++++ |
| Ventricular strain, myocardial mechanics | ++++ | ++ | +++ |
| Inflammation | ++ | ++++ | ++++ |
| Fibrosis/Infiltration | ++ | +++ | ++++ |
| Pericardium/Pericardial effusion | +++ | ++ | ++++ |
| Alternate diagnoses of chest pain syndromes | ++ | +++ | ++++ |
| Cost | Inexpensive | Most expensive | Increasingly affordable; cost-effective |
| Strengths | Portable, widely accessible in most medical institutions, rapid assessment of systolic function and wall motion abnormalities | Highly validated in inflammatory state; molecular and metabolic characterization | High spatial resolution, highly reproducible, volumetric coverage, multi-faceted tissue characterization |
| Limitations | Dependence on optimal acoustic window, nonspecific findings | Exposure to ionizing radiation with use of nuclear tracers, special preparation required | Specific Hardware/software requirements with fewer centers of excellence available; claustrophobia, use of gadolinium contrast |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čiháková, D.; Shi, Y.; Adhikari, B.; Bandettini, W.P.; Cunningham, M.W.; Danthi, N.; Friedrich, M.G.; Liu, P.; Longacre, L.S.; Mann, D.L.; et al. Meeting the Challenges of Myocarditis: New Opportunities for Prevention, Detection, and Intervention—A Report from the 2021 National Heart, Lung, and Blood Institute Workshop. J. Clin. Med. 2022, 11, 5721. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11195721
Čiháková D, Shi Y, Adhikari B, Bandettini WP, Cunningham MW, Danthi N, Friedrich MG, Liu P, Longacre LS, Mann DL, et al. Meeting the Challenges of Myocarditis: New Opportunities for Prevention, Detection, and Intervention—A Report from the 2021 National Heart, Lung, and Blood Institute Workshop. Journal of Clinical Medicine. 2022; 11(19):5721. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11195721
Chicago/Turabian StyleČiháková, Daniela, Yang Shi, Bishow Adhikari, W. Patricia Bandettini, Madeleine W. Cunningham, Narasimhan Danthi, Matthias G. Friedrich, Peter Liu, Lisa Schwartz Longacre, Douglas L. Mann, and et al. 2022. "Meeting the Challenges of Myocarditis: New Opportunities for Prevention, Detection, and Intervention—A Report from the 2021 National Heart, Lung, and Blood Institute Workshop" Journal of Clinical Medicine 11, no. 19: 5721. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11195721