
Intestinal Inflammation and Alterations in the Gut Microbiota in Cystic Fibrosis: A Review of the Current Evidence, Pathophysiology and Future Directions

 and
and 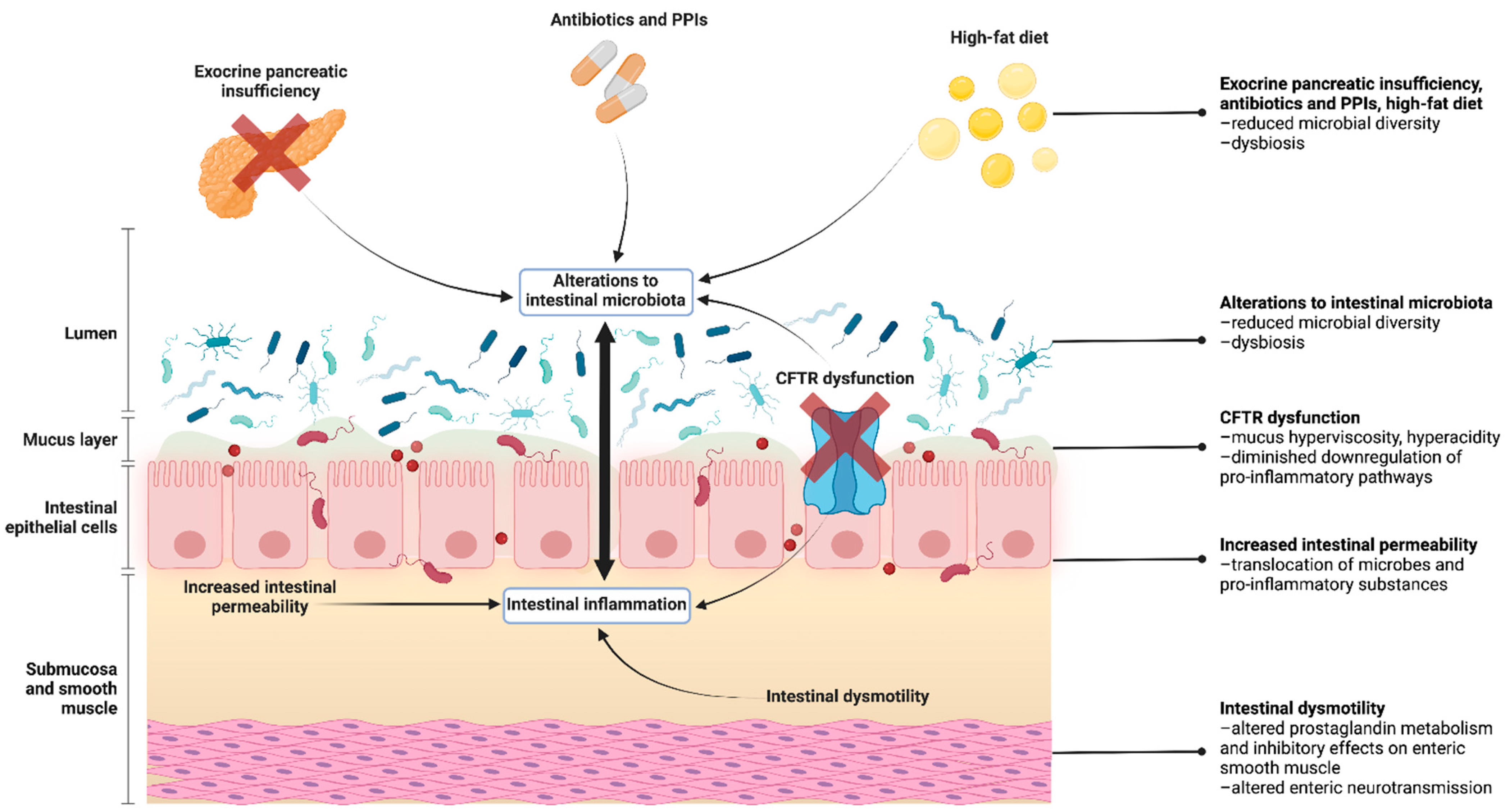{kind=link}
Abstract
:1. Introduction
2. CFTR in the Gastrointestinal Tract
3. Intestinal Inflammation
3.1. The Intestine Is a Site of Inflammation in CF
3.2. Pathogenesis of Intestinal Inflammation in CF
3.2.1. CFTR Dysfunction
3.2.2. Intestinal Dysmotility
3.2.3. Intestinal Dysbiosis
3.2.4. Increased Intestinal Permeability
3.3. Clinical Correlations with Intestinal Inflammation
3.3.1. Exocrine Pancreatic Status, Age, and Lung Function
3.3.2. Growth Parameters
3.3.3. Quality of Life and Hospitalisations
3.3.4. Iatrogenic Factors: High-Fat Diet and Antibiotic Use
4. The CF Gut Microbiome
4.1. Species Diversity and Microbiome Maturation
4.2. Microbial Composition and Functionality
4.3. Pathogenesis of Intestinal Dysbiosis
4.3.1. CFTR Dysfunction
4.3.2. Exocrine Pancreatic Status
4.3.3. Antibiotic and Proton Pump Inhibitor Use
4.3.4. High-Fat Diet
4.4. Clinical Significance of the Gut Microbiome in CF
4.4.1. Pulmonary Function and the Gut–Lung Axis
4.4.2. Growth and Nutritional Status
5. Linking Intestinal Inflammation and Gut Dysbiosis
6. CF Intestinal Disease in the Era of CFTR Modulator Therapies
7. Future Directions
7.1. Probiotics
7.2. The Increased Risk of GI Malignancies
7.3. The Intestinal Virome and Mycobiome
7.4. Multi-Omics Research
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cutting, G. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotnala, S.; Dhasmana, A.; Kashyap, V.K.; Chauhan, S.C.; Yallapu, M.M.; Jaggi, M. A bird eye view on cystic fibrosis: An underestimated multifaceted chronic disorder. Life Sci. 2021, 268, 118959. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.M.; Bonk, M.; Hadjiliadis, D. Cystic Fibrosis: Emerging Understanding and Therapies. Annu. Rev. Med. 2019, 70, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Cystic Fibrosis Mutation Database (CFTR1). 2011. Available online: http://www.genet.sickkids.on.ca/ (accessed on 24 November 2021).
- Sharma, N.; Cutting, G. The genetics and genomics of cystic fibrosis. J. Cyst. Fibros. 2019, 19 (Suppl. 1), S5–S9. [Google Scholar] [CrossRef] [Green Version]
- Ooi, C.Y.; Durie, P.R. Cystic fibrosis from the gastroenterologist’s perspective. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 175–185. [Google Scholar] [CrossRef]
- Dos Santos, A.L.M.; Santos, H.d.; Nogueira, M.B.; Távora, H.T.O.; da Cunha, M.d.J.P.; Seixas, R.B.P.d.; Monte, L.d.V.; de Carvalho, E. Cystic Fibrosis: Clinical Phenotypes in Children and Adolescents. Pediatr. Gastroenterol. Hepatol. Nutr. 2018, 21, 306–314. [Google Scholar] [CrossRef]
- Castellani, C.; Assael, B.M. Cystic fibrosis: A clinical view. Cell. Mol. Life Sci. 2017, 74, 129–140. [Google Scholar] [CrossRef]
- Singh, V.K.; Schwarzenberg, S.J. Pancreatic insufficiency in Cystic Fibrosis. J. Cyst. Fibros. 2017, 16 (Suppl. 2), S70–S78. [Google Scholar] [CrossRef] [Green Version]
- Freeman, A.J.; Ooi, C. Pancreatitis and pancreatic cystosis in Cystic Fibrosis. J. Cyst. Fibros. 2017, 16 (Suppl. 2), S79–S86. [Google Scholar] [CrossRef] [Green Version]
- Ooi, C.Y.; Dorfman, R.; Cipolli, M.; Gonska, T.; Castellani, C.; Keenan, K.; Freedman, S.D.; Zielenski, J.; Berthiaume, Y.; Corey, M.; et al. Type of CFTR Mutation Determines Risk of Pancreatitis in Patients With Cystic Fibrosis. Gastroenterology 2011, 140, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Coffey, M.J.; Ooi, C.Y. Cystic Fibrosis-related Liver Disease is Associated with Increased Disease Burden and Endocrine Comorbidities. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Stonebraker, J.R.; Ooi, C.; Pace, R.G.; Corvol, H.; Knowles, M.R.; Durie, P.R.; Ling, S. Features of Severe Liver Disease With Portal Hypertension in Patients With Cystic Fibrosis. Clin. Gastroenterol. Hepatol. 2016, 14, 1207–1215.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flass, T.; Narkewicz, M.R. Cirrhosis and other liver disease in cystic fibrosis. J. Cyst. Fibros. 2013, 12, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Parisi, G.F.; Di Dio, G.; Franzonello, C.; Gennaro, A.; Rotolo, N.; Lionetti, E.; Leonardi, S. Liver Disease in Cystic Fibrosis: An Update. Zahedan J. Res. Med Sci. 2013, 13, e11215. [Google Scholar] [CrossRef] [Green Version]
- Moran, A.; Dunitz, J.; Nathan, B.; Saeed, A.; Holme, B.; Thomas, W. Cystic fibrosis-related diabetes: Current trends in prevalence, incidence, and mortality. Diabetes Care 2009, 32, 1626–1631. [Google Scholar] [CrossRef] [Green Version]
- Kelsey, R.; Koivula, F.N.M.; McClenaghan, N.H.; Kelly, C. Cystic Fibrosis–Related Diabetes: Pathophysiology and Therapeutic Challenges. Clin. Med. Insights Endocrinol. Diabetes 2019, 12, 1179551419851770. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.C.; Casella, J.L.; Litvin, M.; Dobs, A.S. Male reproductive health in cystic fibrosis. J. Cyst. Fibros. 2019, 18 (Suppl. 2), S105–S110. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Ruan, Y.C.; Xu, W.M.; Chen, J.; Chan, H.C. Regulation of male fertility by CFTR and implications in male infertility. Hum. Reprod. Updat. 2012, 18, 703–713. [Google Scholar] [CrossRef] [Green Version]
- Yamada, A.; Komaki, Y.; Komaki, F.; Micic, D.; Zullow, S.; Sakuraba, A. Risk of gastrointestinal cancers in patients with cystic fibrosis: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 758–767. [Google Scholar] [CrossRef]
- Maisonneuve, P.; Marshall, B.C.; Knapp, E.A.; Lowenfels, A.B. Cancer Risk in Cystic Fibrosis: A 20-Year Nationwide Study from the United States. JNCI J. Natl. Cancer Inst. 2012, 105, 122–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayama, G.; Priya, S.; Niccum, D.E.; Khoruts, A.; Blekhman, R. Interactions between the gut microbiome and host gene regulation in cystic fibrosis. Genome Med. 2020, 12, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowbotham, N.J.; Smith, S.; Leighton, P.A.; Rayner, O.C.; Gathercole, K.; Elliott, Z.C.; Nash, E.F.; Daniels, T.; Duff, A.J.A.; Collins, S.; et al. The top 10 research priorities in cystic fibrosis developed by a partnership between people with CF and healthcare providers. Thorax 2018, 73, 388–390. [Google Scholar] [CrossRef] [Green Version]
- De Palma, F.D.E.; Raia, V.; Kroemer, G.; Maiuri, M.C. The Multifaceted Roles of MicroRNAs in Cystic Fibrosis. Diagnostics 2020, 10, 1102. [Google Scholar] [CrossRef] [PubMed]
- Kalin, N.; Claass, A.; Sommer, M.; Puchelle, E.; Tummler, B. DeltaF508 CFTR protein expression in tissues from patients with cystic fibrosis. J. Clin. Investig. 1999, 103, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- De Lisle, R.C.; Borowitz, D. The cystic fibrosis intestine. Cold Spring Harb. Perspect. Med. 2013, 3, a009753. [Google Scholar] [CrossRef] [Green Version]
- Jakab, R.L.; Collaco, A.M.; Ameen, N.A. Physiological relevance of cell-specific distribution patterns of CFTR, NKCC1, NBCe1, and NHE3 along the crypt-villus axis in the intestine. Am. J. Physiol. Liver Physiol. 2011, 300, G82–G98. [Google Scholar] [CrossRef] [Green Version]
- Venkatasubramanian, J.; Ao, M.; Rao, M.C. Ion transport in the small intestine. Curr. Opin. Gastroenterol. 2010, 26, 123–128. [Google Scholar] [CrossRef]
- Liou, T.G. The Clinical Biology of Cystic Fibrosis Transmembrane Regulator Protein: Its Role and Function in Extrapulmonary Disease. Chest 2019, 155, 605–616. [Google Scholar] [CrossRef]
- Scott, P.; Anderson, K.; Singhania, M.; Cormier, R. Cystic Fibrosis, CFTR, and Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 2891. [Google Scholar] [CrossRef] [Green Version]
- Smyth, R.L.; Croft, N.M.; O’Hea, U.; Marshall, T.G.; Ferguson, A. Intestinal inflammation in cystic fibrosis. Arch. Dis. Child. 2000, 82, 394–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raia, V.; Maiuri, L.; de Ritis, G.; de Vizia, B.; Vacca, L.; Conte, R.; Auricchio, S.; Londei, M. Evidence of Chronic Inflammation in Morphologically Normal Small Intestine of Cystic Fibrosis Patients. Pediatr. Res. 2000, 47, 344–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brecelj, J.; Zidar, N.; Jeruc, J.; Orel, R. Morphological and Functional Assessment of Oesophageal Mucosa Integrity in Children With Cystic Fibrosis. J. Pediatr. Gastroenterol. Nutr. 2016, 62, 757–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werlin, S.L.; Benuri-Silbiger, I.; Kerem, E.; Adler, S.N.; Goldin, E.; Zimmerman, J.; Malka, N.; Cohen, L.; Armoni, S.; Yatzkan-Israelit, Y.; et al. Evidence of Intestinal Inflammation in Patients With Cystic Fibrosis. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 304–308. [Google Scholar] [CrossRef]
- Flass, T.; Tong, S.; Frank, D.N.; Wagner, B.; Robertson, C.; Kotter, C.V.; Sokol, R.J.; Zemanick, E.; Accurso, F.; Hoffenberg, E.; et al. Intestinal Lesions Are Associated with Altered Intestinal Microbiome and Are More Frequent in Children and Young Adults with Cystic Fibrosis and Cirrhosis. PLoS ONE 2015, 10, e0116967. [Google Scholar] [CrossRef] [Green Version]
- Bruzzese, E.; Raia, V.; Gaudiello, G.; Polito, G.; Buccigrossi, V.; Formicola, V.; Guarino, A. Intestinal inflammation is a frequent feature of cystic fibrosis and is reduced by probiotic administration. Aliment. Pharmacol. Ther. 2004, 20, 813–819. [Google Scholar] [CrossRef]
- Rumman, N.; Sultan, M.; El-Chammas, K.; Goh, V.; Salzman, N.; Quintero, D.; Werlin, S. Calprotectin in Cystic Fibrosis. BMC Pediatr. 2014, 14, 133. [Google Scholar] [CrossRef] [Green Version]
- Adriaanse, M.P.M.; Van Der Sande, L.J.T.M.; Neucker, A.M.V.D.; Menheere, P.P.C.A.; Dompeling, E.; Buurman, W.A.; Vreugdenhil, A.C.E. Evidence for a Cystic Fibrosis Enteropathy. PLoS ONE 2015, 10, e0138062. [Google Scholar] [CrossRef] [Green Version]
- Ellemunter, H.; Engelhardt, A.; Schüller, K.; Steinkamp, G. Fecal Calprotectin in Cystic Fibrosis and Its Relation to Disease Parameters: A Longitudinal Analysis for 12 Years. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 438–442. [Google Scholar] [CrossRef]
- Bruzzese, E.; Callegari, M.L.; Raia, V.; Viscovo, S.; Scotto, R.; Ferrari, S.; Morelli, L.; Buccigrossi, V.; Vecchio, A.L.; Ruberto, E.; et al. Disrupted Intestinal Microbiota and Intestinal Inflammation in Children with Cystic Fibrosis and Its Restoration with Lactobacillus GG: A Randomised Clinical Trial. PLoS ONE 2014, 9, e87796. [Google Scholar] [CrossRef] [Green Version]
- Parisi, G.F.; Papale, M.; Rotolo, N.; Aloisio, D.; Tardino, L.; Scuderi, M.G.; Di Benedetto, V.; Nenna, R.; Midulla, F.; Leonardi, S. Severe disease in Cystic Fibrosis and fecal calprotectin levels. Immunobiology 2017, 222, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, F.; Federici, S.; Ferrari, S.; Minuti, A.; Rebecchi, A.; Bruzzese, E.; Buccigrossi, V.; Guarino, A.; Callegari, M.L. Impact of cystic fibrosis disease on archaea and bacteria composition of gut microbiota. FEMS Microbiol. Ecol. 2017, 93, fiw230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Freitas, M.B.; Moreira, E.A.M.; Tomio, C.; Moreno, Y.M.F.; Daltoe, F.P.; Barbosa, E.; Neto, N.L.; Buccigrossi, V.; Guarino, A. Altered intestinal microbiota composition, antibiotic therapy and intestinal inflammation in children and adolescents with cystic fibrosis. PLoS ONE 2018, 13, e0198457. [Google Scholar] [CrossRef] [PubMed]
- Sathe, M.; Huang, R.; Heltshe, S.L.; Eng, A.; Borenstein, E.; Miller, S.I.; Hoffman, L.; Gelfond, D.; Leung, D.H.; Borowitz, D.; et al. Gastrointestinal Factors Associated With Hospitalization in Infants With Cystic Fibrosis: Results from the BONUS Study. J. Pediatr. Gastroenterol. Nutr. 2021, 73, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Enaud, R.; Hooks, K.B.; Barre, A.; Barnetche, T.; Hubert, C.; Massot, M.; Bazin, T.; Clouzeau, H.; Bui, S.; Fayon, M.; et al. Intestinal Inflammation in Children with Cystic Fibrosis Is Associated with Crohn’s-Like Microbiota Disturbances. J. Clin. Med. 2019, 8, 645. [Google Scholar] [CrossRef] [Green Version]
- Beaufils, F.; Mas, E.; Mittaine, M.; Addra, M.; Fayon, M.; Delhaes, L.; Clouzeau, H.; Galode, F.; Lamireau, T.; Bui, S.; et al. Increased Fecal Calprotectin is Associated with Worse Gastrointestinal Symptoms and Quality of Life Scores in Children with Cystic Fibrosis. J. Clin. Med. 2020, 9, 4080. [Google Scholar] [CrossRef]
- Safe, M.; Gifford, A.; Jaffe, A.; Ooi, C.Y. Resolution of Intestinal Histopathology Changes in Cystic Fibrosis after Treatment with Ivacaftor. Ann. Am. Thorac. Soc. 2016, 13, 297–298. [Google Scholar] [CrossRef]
- Davidson, F.; Lock, R.J. Paediatric reference ranges for faecal calprotectin: A UK study. Ann. Clin. Biochem. 2017, 54, 214–218. [Google Scholar] [CrossRef]
- Lin, J.-F.; Chen, J.; Zuo, J.; Yu, A.; Xiao, Z.; Deng, F.; Nie, B.; Jiang, B. Meta-analysis: Fecal calprotectin for assessment of inflammatory bowel disease activity. Inflamm. Bowel. Dis. 2014, 20, 1407–1415. [Google Scholar] [CrossRef]
- Dhaliwal, J.; Leach, S.; Katz, T.; Nahidi, L.; Pang, T.; Lee, J.; Strachan, R.; Day, A.S.; Jaffe, A.; Ooi, C.Y. Intestinal Inflammation and Impact on Growth in Children With Cystic Fibrosis. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Jung, D.; Dong, K.; Jang, J.; Lam, G.Y.; Wilcox, P.G.; Quon, B.S. Circulating CRP and calprotectin to diagnose CF pulmonary exacerbations. J. Cyst. Fibros. 2021, 20, 46–49. [Google Scholar] [CrossRef]
- Wiecek, S.; Wos, H.; Kordys-Darmolińska, B.; Sankiewicz-Szkółka, M.; Grzybowska-Chlebowczyk, U. The concentration of calprotectin in the stools of children with diagnosed cystic fibrosis. Gastroenterol. Rev. 2017, 12, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Shoki, A.H.; Mayer-Hamblett, N.; Wilcox, P.G.; Sin, D.D.; Quon, B.S. Systematic Review of Blood Biomarkers in Cystic Fibrosis Pulmonary Exacerbations. Chest 2013, 144, 1659–1670. [Google Scholar] [CrossRef] [PubMed]
- Tabori, H.; Arnold, C.; Jaudszus, A.; Mentzel, H.-J.; Renz, D.M.; Reinsch, S.; Lorenz, M.; Michl, R.; Gerber, A.; Lehmann, T.; et al. Abdominal symptoms in cystic fibrosis and their relation to genotype, history, clinical and laboratory findings. PLoS ONE 2017, 12, e0174463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolia, R.; Ooi, C.Y.; Lewindon, P.; Bishop, J.; Ranganathan, S.; Harrison, J.; Ford, K.; Van Der Haak, N.; Oliver, M.R. Practical approach to the gastrointestinal manifestations of cystic fibrosis. J. Paediatr. Child Health. 2018, 54, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Ooi, C.Y. The Enigmatic Gut in Cystic Fibrosis: Linking Inflammation, Dysbiosis, and the Increased Risk of Malignancy. Curr. Gastroenterol. Rep. 2017, 19, 6. [Google Scholar] [CrossRef]
- Munck, A. Cystic fibrosis: Evidence for gut inflammation. Int. J. Biochem. Cell Biol. 2014, 52, 180–183. [Google Scholar] [CrossRef]
- Vij, N.; Mazur, S.; Zeitlin, P.L. CFTR is a negative regulator of NFkappaB mediated innate immune response. PLoS ONE 2009, 4, e4664. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, X.; Zhang, J.T.; Tsang, L.L.; Jiang, X.; Chan, H.C. Defective CFTR- beta-catenin interaction promotes NF-kappaB nuclear translocation and intestinal inflammation in cystic fibrosis. Oncotarget 2016, 7, 64030–64042. [Google Scholar] [CrossRef]
- Crites, K.S.-M.; Morin, G.; Orlando, V.; Patey, N.; Cantin, C.; Martel, J.; Brochiero, E.; Mailhot, G. CFTR Knockdown induces proinflammatory changes in intestinal epithelial cells. J. Inflamm. 2015, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Than, B.L.N.; Linnekamp, J.F.; Starr, T.; Largaespada, D.A.; Rod, A.; Zhang, Y.; Bruner, V.; Abrahante, J.; Schumann, A.; Luczak, T.; et al. CFTR is a tumor suppressor gene in murine and human intestinal cancer. Oncogene 2016, 35, 4179–4187. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.Y.; Pang, T.; Leach, S.T.; Katz, T.; Day, A.S.; Jaffe, A. Fecal Human beta-Defensin 2 in Children with Cystic Fibrosis: Is There a Diminished Intestinal Innate Immune Response? Dig. Dis. Sci. 2015, 60, 2946–2952. [Google Scholar] [CrossRef] [PubMed]
- Tétard, C.; Mittaine, M.; Bui, S.; Beaufils, F.; Maumus, P.; Fayon, M.; Burgel, P.-R.; Lamireau, T.; Delhaes, L.; Mas, E.; et al. Reduced Intestinal Inflammation with Lumacaftor/Ivacaftor in Adolescents with Cystic Fibrosis. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.Y.; Syed, S.A.; Rossi, L.; Garg, M.; Needham, B.; Avolio, J.; Young, K.; Surette, M.G.; Gonska, T. Impact of CFTR modulation with Ivacaftor on Gut Microbiota and Intestinal Inflammation. Sci. Rep. 2018, 8, 17834. [Google Scholar] [CrossRef] [Green Version]
- Stallings, V.A.; Sainath, N.; Oberle, M.; Bertolaso, C.; Schall, J.I. Energy Balance and Mechanisms of Weight Gain with Ivacaftor Treatment of Cystic Fibrosis Gating Mutations. J. Pediatr. 2018, 201, 229–237.e4. [Google Scholar] [CrossRef] [PubMed]
- Malagelada, C.; Bendezú, R.; Seguí, S.; Vitrià, J.; Merino, X.; Nieto, A.; Sihuay, D.; Accarino, A.; Molero, X.; Azpiroz, F. Motor dysfunction of the gut in cystic fibrosis. Neurogastroenterol. Motil. 2020, 32, e13883. [Google Scholar] [CrossRef]
- Henen, S.; Denton, C.; Teckman, J.; Borowitz, D.; Patel, D. Review of Gastrointestinal Motility in Cystic Fibrosis. J. Cyst. Fibros. 2021, 20, 578–585. [Google Scholar] [CrossRef]
- De Lisle, R.C.; Sewell, R.; Meldi, L. Enteric circular muscle dysfunction in the cystic fibrosis mouse small intestine. Neurogastroenterol. Motil. 2010, 22, 341-e87. [Google Scholar] [CrossRef] [Green Version]
- De Lisle, R.C.; Meldi, L.; Roach, E.; Flynn, M.; Sewell, R. Mast Cells and Gastrointestinal Dysmotility in the Cystic Fibrosis Mouse. PLoS ONE 2009, 4, e4283. [Google Scholar] [CrossRef] [Green Version]
- Risse, P.-A.; Kachmar, L.; Matusovsky, O.S.; Novali, M.; Gil, F.R.; Javeshghani, S.; Keary, R.; Haston, C.K.; Michoud, M.-C.; Martin, J.G.; et al. Ileal smooth muscle dysfunction and remodeling in cystic fibrosis. Am. J. Physiol. Liver Physiol. 2012, 303, G1–G8. [Google Scholar] [CrossRef] [Green Version]
- Hedsund, C.; Gregersen, T.; Joensson, I.M.; Olesen, H.V.; Krogh, K. Gastrointestinal transit times and motility in patients with cystic fibrosis. Scand. J. Gastroenterol. 2012, 47, 920–926. [Google Scholar] [CrossRef]
- Ng, C.; Dellschaft, N.S.; Hoad, C.L.; Marciani, L.; Ban, L.; Prayle, A.P.; Barr, H.L.; Jaudszus, A.; Mainz, J.G.; Spiller, R.C.; et al. Postprandial changes in gastrointestinal function and transit in cystic fibrosis assessed by Magnetic Resonance Imaging. J. Cyst. Fibros. 2021, 20, 591–597. [Google Scholar] [CrossRef] [PubMed]
- De Lisle, R.C.; Meldi, L.; Flynn, M.; Jansson, K. Altered Eicosanoid Metabolism in the Cystic Fibrosis Mouse Small Intestine. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Gu, H.; Qiu, Y.; Guo, Y.; Korteweg, C.; Huang, J.; Gu, J. Expression of Cystic Fibrosis Transmembrane Conductance Regulator in Ganglia of Human Gastrointestinal Tract. Sci. Rep. 2016, 6, 30926. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.M.; Johansson, O.; Le, H.; Rao, K.; Markus, I.; Perera, D.S.; Lubowski, D.Z.; King, D.W.; Zhang, L.; Chen, H.; et al. Cystic fibrosis transmembrane conductance regulator modulates enteric cholinergic activities and is abnormally expressed in the enteric ganglia of patients with slow transit constipation. J. Gastroenterol. 2019, 54, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Coffey, M.J.; Nielsen, S.; Wemheuer, B.; Kaakoush, N.O.; Garg, M.; Needham, B.; Pickford, R.; Jaffe, A.; Thomas, T.; Ooi, C.Y. Gut Microbiota in Children With Cystic Fibrosis: A Taxonomic and Functional Dysbiosis. Sci. Rep. 2019, 9, 18593. [Google Scholar] [CrossRef]
- Hoffman, L.R.; Pope, C.E.; Hayden, H.S.; Heltshe, S.; Levy, R.; McNamara, S.; Jacobs, M.A.; Rohmer, L.; Radey, M.; Ramsey, B.W.; et al. Escherichia coli dysbiosis correlates with gastrointestinal dysfunction in children with cystic fibrosis. Clin. Infect. Dis. 2014, 58, 396–399. [Google Scholar] [CrossRef] [Green Version]
- Fallahi, G.; Motamed, F.; Yousefi, A.; Shafieyoun, A.; Najafi, M.; Khodadad, A.; Farhmand, F.; Ahmadvand, A.; Rezaei, N. The effect of probiotics on fecal calprotectin in patients with cystic fibrosis. Turk. J. Pediatr. 2013, 55, 475–478. [Google Scholar]
- del Campo, R.; Garriga, M.; Pérez-Aragón, A.; Guallarte, P.; Lamas, A.; Máiz, L.; Bayón, C.; Roy, G.; Cantón, R.; Zamora, J.; et al. Improvement of digestive health and reduction in proteobacterial populations in the gut microbiota of cystic fibrosis patients using a Lactobacillus reuteri probiotic preparation: A double blind prospective study. J. Cyst. Fibros. 2014, 13, 716–722. [Google Scholar] [CrossRef] [Green Version]
- Coffey, M.J.; Garg, M.; Homaira, N.; Jaffe, A.; Ooi, C.Y. Probiotics for people with cystic fibrosis. Cochrane Database Syst. Rev. 2020, 1, CD012949. [Google Scholar] [CrossRef]
- Fukui, H. Increased Intestinal Permeability and Decreased Barrier Function: Does It Really Influence the Risk of Inflammation? Inflamm. Intest. Dis. 2016, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- De Lisle, R.C. Disrupted tight junctions in the small intestine of cystic fibrosis mice. Cell Tissue Res. 2014, 355, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lisle, R.C.; Mueller, R.; Boyd, M. Impaired Mucosal Barrier Function in the Small Intestine of the Cystic Fibrosis Mouse. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallberg, K.; Grzegorczyk, A.; Larson, G.; Strandvik, B. Intestinal Permeability in Cystic Fibrosis in Relation to Genotype. J. Pediatr. Gastroenterol. Nutr. 1997, 25, 290–295. [Google Scholar] [CrossRef]
- van Elburg, R.M.; Uil, J.J.; van Aalderen, W.M.; Mulder, C.J.; Heymans, H.S. Intestinal permeability in exocrine pancreatic insufficiency due to cystic fibrosis or chronic pancreatitis. Pediatr. Res. 1996, 39, 985–991. [Google Scholar] [CrossRef] [Green Version]
- Mack, D.R.; Flick, J.A.; Durie, P.R.; Rosenstein, B.J.; Ellis, L.E.; Perman, J.A. Correlation of intestinal lactulose permeability with exocrine pancreatic dysfunction. J. Pediatr. 1992, 120, 696–701. [Google Scholar] [CrossRef]
- Dalzell, A.M.; Freestone, N.S.; Billington, D.; Heaf, D.P. Small intestinal permeability and orocaecal transit time in cystic fibrosis. Arch. Dis. Child. 1990, 65, 585–588. [Google Scholar] [CrossRef] [Green Version]
- Mankertz, J.; Schulzke, J.-D. Altered permeability in inflammatory bowel disease: Pathophysiology and clinical implications. Curr. Opin. Gastroenterol. 2007, 23, 379–383. [Google Scholar] [CrossRef]
- Hering, N.A.; Fromm, M.; Schulzke, J.D. Determinants of colonic barrier function in inflammatory bowel disease and potential therapeutics. J. Physiol. 2012, 590, 1035–1044. [Google Scholar] [CrossRef]
- Turpin, W.; Lee, S.H.; Raygoza Garay, J.A.; Madsen, K.L.; Meddings, J.B.; Bedrani, L.; Power, N.; Espin-Garcia, O.; Xu, W.; Smith, M.I.; et al. Increased Intestinal Permeability Is Associated With Later Development of Crohn’s Disease. Gastroenterology 2020, 159, 2092–2100.e5. [Google Scholar] [CrossRef]
- Ahmad, R.; Sorrell, M.F.; Batra, S.K.; Dhawan, P.; Singh, A.B. Gut permeability and mucosal inflammation: Bad, good or context dependent. Mucosal Immunol. 2017, 10, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, M.; Leach, S.T.; Coffey, M.J.; Katz, T.; Strachan, R.; Pang, T.; Needham, B.; Lui, K.; Ali, F.; Day, A.S.; et al. Age-dependent variation of fecal calprotectin in cystic fibrosis and healthy children. J. Cyst. Fibros. 2017, 16, 631–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, M.; Leach, S.T.; Pang, T.; Needham, B.; Coffey, M.J.; Katz, T.; Strachan, R.; Widger, J.; Field, P.; Belessis, Y.; et al. Age-related levels of fecal M2-pyruvate kinase in children with cystic fibrosis and healthy children 0 to 10 years old. J. Cyst. Fibros. 2018, 17, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxby, N.P.C.; Kench, A.; King, S.; Crowder, T.; van der Haak, N. The Australian and New Zealand Cystic Fibrosis Nutrition Guideline Authorship Group. Nutrition Guidelines for Cystic Fibrosis in Australia and New Zealand; Bell, S.C., Ed.; Thoracic Society of Australia and New Zealand: Sydney, Australia, 2017. [Google Scholar]
- Smith, C.; Winn, A.; Seddon, P.; Ranganathan, S. A fat lot of good: Balance and trends in fat intake in children with cystic fibrosis. J. Cyst. Fibros. 2012, 11, 154–157. [Google Scholar] [CrossRef] [Green Version]
- Woestenenk, J.W.; Schulkes, D.A.; Schipper, H.S.; van der Ent, C.K.; Houwen, R.H. Dietary intake and lipid profile in children and adolescents with cystic fibrosis. J. Cyst. Fibros. 2017, 16, 410–417. [Google Scholar] [CrossRef]
- Woestenenk, J.; Castelijns, S.; Van Der Ent, C.; Houwen, R. Dietary intake in children and adolescents with cystic fibrosis. Clin. Nutr. 2014, 33, 528–532. [Google Scholar] [CrossRef]
- McDonald, C.M.; Bowser, E.K.; Farnham, K.; Alvarez, J.A.; Padula, L.; Rozga, M. Dietary Macronutrient Distribution and Nutrition Outcomes in Persons with Cystic Fibrosis: An Evidence Analysis Center Systematic Review. J. Acad. Nutr. Diet. 2021, 121, 1574–1590.e3. [Google Scholar] [CrossRef]
- Calvo-Lerma, J.; Hulst, J.; Boon, M.; Martins, T.; Ruperto, M.; Colombo, C.; Fornés-Ferrer, V.; Woodcock, S.; Claes, I.; Asseiceira, I.; et al. The Relative Contribution of Food Groups to Macronutrient Intake in Children with Cystic Fibrosis: A European Multicenter Assessment. J. Acad. Nutr. Diet. 2019, 119, 1305–1319. [Google Scholar] [CrossRef]
- Sutherland, R.; Katz, T.; Liu, V.; Quintano, J.; Brunner, R.; Tong, C.W.; Collins, C.E.; Ooi, C.Y. Dietary intake of energy-dense, nutrient-poor and nutrient-dense food sources in children with cystic fibrosis. J. Cyst. Fibros. 2018, 17, 804–810. [Google Scholar] [CrossRef]
- Debray, D.; el Mourabit, H.; Merabtene, F.; Brot, L.; Ulveling, D.; Chrétien, Y.; Rainteau, D.; Moszer, I.; Wendum, D.; Sokol, H.; et al. Diet-Induced Dysbiosis and Genetic Background Synergize with Cystic Fibrosis Transmembrane Conductance Regulator Deficiency to Promote Cholangiopathy in Mice. Hepatol. Commun. 2018, 2, 1533–1549. [Google Scholar] [CrossRef]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.J.; Magness, S.; Jobin, C.; Lund, P.K. High-Fat Diet: Bacteria Interactions Promote Intestinal Inflammation Which Precedes and Correlates with Obesity and Insulin Resistance in Mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulhane, M.; Murray, L.; Lourie, R.; Tong, H.; Sheng, Y.H.; Wang, R.; Kang, A.; Schreiber, V.; Wong, K.Y.; Magor, G.; et al. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation that is Reversed by IL-22. Sci. Rep. 2016, 6, 28990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, M.; Whisner, C.; Al-Nakkash, L.; Sweazea, K.L. Six-Week High-Fat Diet Alters the Gut Microbiome and Promotes Cecal Inflammation, Endotoxin Production, and Simple Steatosis without Obesity in Male Rats. Lipids 2019, 54, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-A.; Gu, W.; Lee, I.-A.; Joh, E.-H.; Kim, D.-H. High Fat Diet-Induced Gut Microbiota Exacerbates Inflammation and Obesity in Mice via the TLR4 Signaling Pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef]
- Knoop, K.A.; McDonald, K.G.; Kulkarni, D.H.; Newberry, R.D. Antibiotics promote inflammation through the translocation of native commensal colonic bacteria. Gut 2016, 65, 1100–1109. [Google Scholar] [CrossRef] [Green Version]
- Clemente, J.C.; Manasson, J.; Scher, J.U. The role of the gut microbiome in systemic inflammatory disease. BMJ 2018, 360, j5145. [Google Scholar] [CrossRef]
- Ahmed, I.; Roy, B.C.; Khan, S.A.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Duytschaever, G.; Huys, G.; Bekaert, M.; Boulanger, L.; De Boeck, K.; Vandamme, P. Cross-Sectional and Longitudinal Comparisons of the Predominant Fecal Microbiota Compositions of a Group of Pediatric Patients with Cystic Fibrosis and Their Healthy Siblings. Appl. Environ. Microbiol. 2011, 77, 8015–8024. [Google Scholar] [CrossRef] [Green Version]
- Stiemsma, L.T.; Michels, K.B. The Role of the Microbiome in the Developmental Origins of Health and Disease. Pediatrics 2018, 141, e20172437. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Chung, J.; Battaglia, T.; Henderson, N.; Jay, M.; Li, H.; Lieber, A.D.; Wu, F.; Perez-Perez, G.I.; Chen, Y.; et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 2016, 8, 343ra382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, H.S.; Eng, A.; Pope, C.E.; Brittnacher, M.J.; Vo, A.T.; Weiss, E.J.; Hager, K.R.; Martin, B.D.; Leung, D.H.; Heltshe, S.L.; et al. Fecal dysbiosis in infants with cystic fibrosis is associated with early linear growth failure. Nat. Med. 2020, 26, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Vernocchi, P.; del Chierico, F.; Russo, A.; Majo, F. Gut microbiota signatures in cystic fibrosis: Loss of host CFTR function drives the microbiota enterophenotype. PLoS ONE 2018, 13, e0208171. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Needham, B.; Leach, S.T.; Day, A.S.; Jaffe, A.; Thomas, T.; Ooi, C. Disrupted progression of the intestinal microbiota with age in children with cystic fibrosis. Sci. Rep. 2016, 6, 24857. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, M.L.; Limon, J.J.; Bar, A.S.; Leal, C.A.; Gargus, M.; Tang, J.; Brown, J.; Funari, V.A.; Wang, H.L.; Crother, T.; et al. Immunological Consequences of Intestinal Fungal Dysbiosis. Cell Host Microbe 2016, 19, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Huffnagle, G.B.; Noverr, M. The emerging world of the fungal microbiome. Trends Microbiol. 2013, 21, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Fragkou, P.C.; Karaviti, D.; Zemlin, M.; Skevaki, C. Impact of Early Life Nutrition on Children’s Immune System and Noncommunicable Diseases Through Its Effects on the Bacterial Microbiome, Virome and Mycobiome. Front. Immunol. 2021, 12, 644269. [Google Scholar] [CrossRef]
- Thavamani, A.; Salem, I.; Sferra, T.J.; Sankararaman, S. Impact of Altered Gut Microbiota and Its Metabolites in Cystic Fibrosis. Metabolites 2021, 11, 123. [Google Scholar] [CrossRef]
- Valdes, A.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Burke, D.; Fouhy, F.; Harrison, M.J.; Rea, M.C.; Cotter, P.D.; O’Sullivan, O.; Stanton, C.; Hill, C.; Shanahan, F.; Plant, B.J.; et al. The altered gut microbiota in adults with cystic fibrosis. BMC Microbiol. 2017, 17, 58. [Google Scholar]
- Nobel, Y.R.; Cox, L.M.; Kirigin, F.F.; Bokulich, N.A.; Yamanishi, S.; Teitler, I.; Chung, J.; Sohn, J.; Barber, C.M.; Goldfarb, D.S.; et al. Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat. Commun. 2015, 6, 7486. [Google Scholar] [CrossRef]
- Kristensen, M.; Prevaes, S.M.; Kalkman, G.; Tramper-Stranders, G.A.; Hasrat, R.; de Winter-de Groot, K.M.; Janssens, H.M.; Tiddens, H.A.; van Westreenen, M.; Sanders, E.A.; et al. Development of the gut microbiota in early life: The impact of cystic fibrosis and antibiotic treatment. J. Cyst. Fibros. 2020, 19, 553–561. [Google Scholar] [CrossRef]
- Antosca, K.M.; Chernikova, D.A.; Price, C.E.; Ruoff, K.L.; Li, K.; Guill, M.F.; Sontag, N.R.; Morrison, H.G.; Hao, S.; Drumm, M.L.; et al. Altered Stool Microbiota of Infants with Cystic Fibrosis Shows a Reduction in Genera Associated with Immune Programming from Birth. J. Bacteriol. 2019, 201, e00274-19. [Google Scholar] [CrossRef] [Green Version]
- Debyser, G.; Mesuere, B.; Clement, L.; Van de Weygaert, J.; Van Hecke, P.; Duytschaever, G.; Aerts, M.; Dawyndt, P.; De Boeck, K.; Vandamme, P.; et al. Faecal proteomics: A tool to investigate dysbiosis and inflammation in patients with cystic fibrosis. J. Cyst. Fibros. 2016, 15, 242–250. [Google Scholar] [CrossRef] [Green Version]
- Matamouros, S.; Hayden, H.S.; Hager, K.R.; Brittnacher, M.J.; Lachance, K.; Weiss, E.J.; Pope, C.E.; Imhaus, A.-F.; McNally, C.P.; Borenstein, E.; et al. Adaptation of commensal proliferating Escherichia coli to the intestinal tract of young children with cystic fibrosis. Proc. Natl. Acad. Sci. USA 2018, 115, 1605–1610. [Google Scholar] [CrossRef] [Green Version]
- Manor, O.; Levy, R.; Pope, C.E.; Hayden, H.S.; Brittnacher, M.J.; Carr, R.; Radey, M.C.; Hager, K.R.; Heltshe, S.L.; Ramsey, B.W.; et al. Metagenomic evidence for taxonomic dysbiosis and functional imbalance in the gastrointestinal tracts of children with cystic fibrosis. Sci. Rep. 2016, 6, 22493. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Leong, L.E.; Keating, R.L.; Kanno, T.; Abell, G.C.; Mobegi, F.M.; Choo, J.M.; Wesselingh, S.L.; Mason, A.J.; Burr, L.D.; et al. Opportunistic bacteria confer the ability to ferment prebiotic starch in the adult cystic fibrosis gut. Gut Microbes 2019, 10, 367–381. [Google Scholar] [CrossRef] [Green Version]
- Meeker, S.M.; Mears, K.S.; Sangwan, N.; Brittnacher, M.J.; Weiss, E.J.; Treuting, P.M.; Tolley, N.; Pope, C.E.; Hager, K.R.; Vo, A.T.; et al. CFTR dysregulation drives active selection of the gut microbiome. PLOS Pathog. 2020, 16, e1008251. [Google Scholar] [CrossRef] [PubMed]
- Schippa, S.; Iebba, V.; Santangelo, F.; Gagliardi, A.; de Biase, R.V.; Stamato, A.; Bertasi, S.; Lucarelli, M.; Conte, M.P.; Quattrucci, S. Cystic fibrosis transmembrane conductance regulator (CFTR) allelic variants relate to shifts in faecal microbiota of cystic fibrosis patients. PLoS ONE 2013, 8, e61176. [Google Scholar] [CrossRef]
- Ritz, S.; Hahn, D.; Wami, H.T.; Tegelkamp, K.; Dobrindt, U.; Schnekenburger, J. Gut microbiome as a response marker for pancreatic enzyme replacement therapy in a porcine model of exocrine pancreas insufficiency. Microb. Cell Factories 2020, 19, 221. [Google Scholar] [CrossRef]
- Keeney, K.M.; Yurist-Doutsch, S.; Arrieta, M.-C.; Finlay, B.B. Effects of Antibiotics on Human Microbiota and Subsequent Disease. Annu. Rev. Microbiol. 2014, 68, 217–235. [Google Scholar] [CrossRef] [PubMed]
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol. 2015, 6, 1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianiro, G.; Tilg, H.; Gasbarrini, A. Antibiotics as deep modulators of gut microbiota: Between good and evil. Gut 2016, 65, 1906–1915. [Google Scholar] [CrossRef] [PubMed]
- Yassour, M.; Vatanen, T.; Siljander, H.; Hämäläinen, A.-M.; Härkönen, T.; Ryhänen, S.J.; Franzosa, E.A.; Vlamakis, H.; Huttenhower, C.; Gevers, D.; et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 2016, 8, 343ra81. [Google Scholar] [CrossRef] [Green Version]
- Jernberg, C.; Lofmark, S.; Edlund, C.; Jansson, J.K. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology 2010, 156, 3216–3223. [Google Scholar] [CrossRef] [Green Version]
- Langdon, A.; Crook, N.; Dantas, G. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 2016, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Loman, B.R.; Shrestha, C.L.; Thompson, R.; Groner, J.A.; Mejias, A.; Ruoff, K.L.; O’Toole, G.A.; Bailey, M.T.; Kopp, B.T. Age and environmental exposures influence the fecal bacteriome of young children with cystic fibrosis. Pediatr. Pulmonol. 2020, 55, 1661–1670. [Google Scholar] [CrossRef]
- Duytschaever, G.; Huys, G.; Bekaert, M.; Boulanger, L.; De Boeck, K.; Vandamme, P. Dysbiosis of bifidobacteria and Clostridium cluster XIVa in the cystic fibrosis fecal microbiota. J. Cyst. Fibros. 2013, 12, 206–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duytschaever, G.; Huys, G.; Boulanger, L.; De Boeck, K.; Vandamme, P. Amoxicillin-clavulanic acid resistance in fecal Enterobacteriaceae from patients with cystic fibrosis and healthy siblings. J. Cyst. Fibros. 2013, 12, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Imhann, F.; Bonder, M.J.; Vila, A.V.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.A.; Cenit, M.C.; Harmsen, H.J.M.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, M.A.; Goodrich, J.K.; Maxan, M.-E.; Freedberg, D.E.; Abrams, J.A.; Poole, A.; Sutter, J.L.; Welter, D.; Ley, R.; Bell, J.; et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut 2016, 65, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Rosen, R.; Hu, L.; Amirault, J.; Khatwa, U.; Ward, D.V.; Onderdonk, A. 16S Community Profiling Identifies Proton Pump Inhibitor Related Differences in Gastric, Lung, and Oropharyngeal Microflora. J. Pediatr. 2015, 166, 917–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M., Jr.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet Dominates Host Genotype in Shaping the Murine Gut Microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Kim, N.; Yoon, H.; Nam, R.H.; Lee, D.H. Microbial Changes and Host Response in F344 Rat Colon Depending on Sex and Age Following a High-Fat Diet. Front. Microbiol. 2018, 9, 2236. [Google Scholar] [CrossRef] [Green Version]
- Anitha, M.; Reichardt, F.; Tabatabavakili, S.; Nezami, B.G.; Chassaing, B.; Mwangi, S.; Vijay-Kumar, M.; Gewirtz, A.; Srinivasan, S. Intestinal Dysbiosis Contributes to the Delayed Gastrointestinal Transit in High-Fat Diet Fed Mice. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Tomas, J.; Mulet, C.; Saffarian, A.; Cavin, J.-B.; Ducroc, R.; Regnault, B.; Tan, C.K.; Duszka, K.; Burcelin, R.; Wahli, W.; et al. High-fat diet modifies the PPAR-gamma pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proc. Natl. Acad. Sci. USA 2016, 113, E5934–E5943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of high-fat diet on gut microbiota: A driving force for chronic disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Enaud, R.; Prevel, R.; Ciarlo, E.; Beaufils, F.; Wieërs, G.; Guery, B.; Delhaes, L. The Gut-Lung Axis in Health and Respiratory Diseases: A Place for Inter-Organ and Inter-Kingdom Crosstalks. Front. Cell. Infect. Microbiol. 2020, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Dang, A.T.; Marsland, B.J. Microbes, metabolites, and the gut-lung axis. Mucosal. Immunol. 2019, 12, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrêa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A.R. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wu, W.; Liu, Z.; Cong, Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J. Gastroenterol. 2017, 52, 1–8. [Google Scholar] [CrossRef]
- Hoen, A.G.; Li, J.; Moulton, L.A.; O’Toole, G.A.; Housman, M.L.; Koestler, D.C.; Guill, M.F.; Moore, J.; Hibberd, P.L.; Morrison, H.; et al. Associations between Gut Microbial Colonization in Early Life and Respiratory Outcomes in Cystic Fibrosis. J. Pediatr. 2015, 167, 138–147.e3. [Google Scholar] [CrossRef] [Green Version]
- Hiippala, K.; Kainulainen, V.; Suutarinen, M.; Heini, T.; Bowers, J.R.; Jasso-Selles, D.; Lemmer, D.; Valentine, M.; Barnes, R.; Engelthaler, D.M.; et al. Isolation of Anti-Inflammatory and Epithelium Reinforcing Bacteroides and Parabacteroides Spp. from A Healthy Fecal Donor. Nutrients 2020, 12, 935. [Google Scholar] [CrossRef] [Green Version]
- Lai, H.C.; Lin, T.; Chen, T.; Kuo, Y.; Chang, C.; Wu, T.; Shu, C.; Tsai, Y.; Swift, S.; Lu, C. Gut microbiota modulates COPD pathogenesis: Role of anti-inflammatory Parabacteroides goldsteinii lipopolysaccharide. Gut 2022, 71, 309–321. [Google Scholar] [CrossRef]
- Sanders, D.B.; Fink, A.; Mayer-Hamblett, N.; Schechter, M.S.; Sawicki, G.S.; Rosenfeld, M.; Flume, P.A.; Morgan, W.J. Early Life Growth Trajectories in Cystic Fibrosis are Associated with Pulmonary Function at Age 6 Years. J. Pediatr. 2015, 167, 1081–1088.e1. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, A.L.; Mannik, L.A.; Walsh, S.; Brotherwood, M.; Robert, R.; Darling, P.B.; Nisenbaum, R.; Moerman, J.; Stanojevic, S. Longitudinal trends in nutritional status and the relation between lung function and BMI in cystic fibrosis: A population-based cohort study. Am. J. Clin. Nutr. 2013, 97, 872–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinkamp, G.; Wiedemann, B. Relationship between nutritional status and lung function in cystic fibrosis: Cross sectional and longitudinal analyses from the German CF quality assurance (CFQA) project. Thorax 2002, 57, 596–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, E.H.; Quinton, H.; Borowitz, D. Better Nutritional Status in Early Childhood Is Associated with Improved Clinical Outcomes and Survival in Patients with Cystic Fibrosis. J. Pediatr. 2013, 162, 530–535.e1. [Google Scholar] [CrossRef] [PubMed]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids from Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Maciejewska, D.; Skonieczna-Żydecka, K.; Łukomska, A.; Gutowska, I.; Dec, K.; Kupnicka, P.; Palma, J.; Pilutin, A.; Marlicz, W.; Stachowska, E. The short chain fatty acids and lipopolysaccharides status in Sprague-Dawley rats fed with high-fat and high-cholesterol diet. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2018, 69, 205–210. [Google Scholar]
- Parada Venegas, D.; de la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Genet. 2021, 19, 77–94. [Google Scholar] [CrossRef]
- Dengler, F.; Kraetzig, A.; Gabel, G. Butyrate Protects Porcine Colon Epithelium from Hypoxia-Induced Damage on a Functional Level. Nutrients 2021, 13, 305. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Kim, B.G.; Kim, J.H.; Chun, J.; Im, J.P.; Kim, J.S. Sodium butyrate inhibits the NF-kappa B signaling pathway and histone deacetylation, and attenuates experimental colitis in an IL-10 independent manner. Int. Immunopharmacol. 2017, 51, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Yap, Y.A.; McLeod, K.H.; McKenzie, C.I.; Gavin, P.G.; Davalos-Salas, M.; Richards, J.L.; Moore, R.J.; Lockett, T.J.; Clarke, J.M.; Eng, V.V.; et al. An acetate-yielding diet imprints an immune and anti-microbial programme against enteric infection. Clin. Transl. Immunol. 2021, 10, e1233. [Google Scholar] [CrossRef]
- Diao, H.; Jiao, A.R.; Yu, B.; Mao, X.B.; Chen, D.W. Gastric infusion of short-chain fatty acids can improve intestinal barrier function in weaned piglets. Genes Nutr. 2019, 14, 4. [Google Scholar] [CrossRef]
- Liu, L.; Sun, D.; Mao, S.; Zhu, W.; Liu, J. Infusion of sodium butyrate promotes rumen papillae growth and enhances expression of genes related to rumen epithelial VFA uptake and metabolism in neonatal twin lambs. J. Anim. Sci. 2019, 97, 909–921. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Wang, F.; Yuan, J.; Li, J.; Jiang, D.; Zhang, J.; Li, H.; Wang, R.; Tang, J.; Huang, T.; et al. Effects of dietary fat on gut microbiota and faecal metabolites, and their relationship with cardiometabolic risk factors: A 6-month randomised controlled-feeding trial. Gut 2019, 68, 1417–1429. [Google Scholar] [CrossRef] [Green Version]
- Holota, Y.; Dovbynchuk, T.; Kaji, I.; Vareniuk, I.; Dzyubenko, N.; Chervinska, T.; Zakordonets, L.; Stetska, V.; Ostapchenko, L.; Serhiychuk, T.; et al. The long-term consequences of antibiotic therapy: Role of colonic short-chain fatty acids (SCFA) system and intestinal barrier integrity. PLoS ONE 2019, 14, e0220642. [Google Scholar] [CrossRef]
- Holota, Y.V.; Holubenko, O.O.; Ostapchuk, A.M.; Serhiychuk, T.M.; Zakordonets, L.V.; Tolstanova, G.M. Fecal short-chain fatty acids at different time points after ceftriaxone administration in rats. Ukr. Biochem. J. 2017, 89, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Guinan, J.; Wang, S.; Hazbun, T.R.; Yadav, H.; Thangamani, S. Antibiotic-induced decreases in the levels of microbial-derived short-chain fatty acids correlate with increased gastrointestinal colonization of Candida albicans. Sci. Rep. 2019, 9, 8872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tottey, W.; Feria-Gervasio, D.; Gaci, N.; Laillet, B.; Pujos, E.; Martin, J.-F.; Sebedio, J.-L.; Sion, B.; Jarrige, J.-F.; Alric, M.; et al. Colonic Transit Time Is a Driven Force of the Gut Microbiota Composition and Metabolism: In Vitro Evidence. J. Neurogastroenterol. Motil. 2017, 23, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roager, H.M.; Hansen, L.B.S.; Bahl, M.I.; Frandsen, H.L.; Carvalho, V.; Gøbel, R.J.; Dalgaard, M.D.; Plichta, D.R.; Sparholt, M.H.; Vestergaard, H.; et al. Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat. Microbiol. 2016, 1, 16093. [Google Scholar] [CrossRef] [PubMed]
- Scales, B.S.; Dickson, R.P.; Huffnagle, G.B. A tale of two sites: How inflammation can reshape the microbiomes of the gut and lungs. J. Leukoc. Biol. 2016, 100, 943–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, S.E.; Baumler, A.J. Dysbiosis in the inflamed intestine: Chance favors the prepared microbe. Gut. Microbes. 2014, 5, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2007, 2, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Imhann, F.; Vila, A.V.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; Van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef]
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory Bowel Disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef]
- Caruso, R.; Lo, B.C.; Núñez, G. Host–microbiota interactions in inflammatory bowel disease. Nat. Rev. Immunol. 2020, 20, 411–426. [Google Scholar] [CrossRef]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical Mechanism of the Cystic Fibrosis Transmembrane Conductance Regulator Potentiator Ivacaftor in G551D-mediated Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelfond, D.; Heltshe, S.; Ma, C.; Rowe, S.M.; Frederick, C.; Uluer, A.; Sicilian, L.; Konstan, M.; Tullis, E.; Roach, C.R.N.; et al. Impact of CFTR Modulation on Intestinal pH, Motility, and Clinical Outcomes in Patients With Cystic Fibrosis and the G551D Mutation. Clin. Transl. Gastroenterol. 2017, 8, e81. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Groot, K.D.W.-D.; Berkers, G.; Chu, M.; Arp, K.; Ghijsen, S.; Heijerman, H.; Arets, H.; Majoor, C.; Janssens, H.; et al. Individual and Group Response of Treatment with Ivacaftor on Airway and Gut Microbiota in People with CF and a S1251N Mutation. J. Pers. Med. 2021, 11, 350. [Google Scholar] [CrossRef]
- Pope, C.; Vo, A.; Hayden, H.; Weiss, E.; Durfey, S.; McNamara, S.; Ratjen, A.; Grogan, B.; Carter, S.; Nay, L.; et al. Changes in fecal microbiota with CFTR modulator therapy: A pilot study. J. Cyst. Fibros. 2021, 20, 742–746. [Google Scholar] [CrossRef]
- Neri, L.D.C.L.; Taminato, M.; da Silva, L.V.R.F. Systematic Review of Probiotics for Cystic Fibrosis Patients: Moving Forward. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Van Biervliet, S.; Declercq, D.; Somerset, S. Clinical effects of probiotics in cystic fibrosis patients: A systematic review. Clin. Nutr. ESPEN 2017, 18, 37–43. [Google Scholar] [CrossRef]
- Anderson, J.L.; Miles, C.; Tierney, A.C. Effect of probiotics on respiratory, gastrointestinal and nutritional outcomes in patients with cystic fibrosis: A systematic review. J. Cyst. Fibros. 2017, 16, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Nikniaz, Z.; Nikniaz, L.; Bilan, N.; Somi, M.H.; Faramarzi, E. Does probiotic supplementation affect pulmonary exacerbation and intestinal inflammation in cystic fibrosis: A systematic review of randomized clinical trials. World J. Pediatr. 2017, 13, 307–313. [Google Scholar] [CrossRef]
- Ananthan, A.; Balasubramanian, H.; Rao, S.; Patole, S. Probiotic supplementation in children with cystic fibrosis—a systematic review. Eur. J. Pediatr. 2016, 175, 1255–1266. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal Inflammation and Cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and Colon Cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef] [PubMed]
- Ekbom, A.; Helmick, C.; Zack, M.; Adami, H.O. Ulcerative colitis and colorectal cancer. A population-based study. N. Engl. J. Med. 1990, 323, 1228–1233. [Google Scholar] [CrossRef]
- Rutter, M.; Saunders, B.; Wilkinson, K.; Rumbles, S.; Schofield, G.; Kamm, M.; Williams, C.; Price, A.; Talbot, I.; Forbes, A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 2004, 126, 451–459. [Google Scholar] [CrossRef]
- Itzkowitz, S.H.; Yio, X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: The role of inflammation. Am. J. Physiol. Gastrointest. Liver. Physiol. 2004, 287, G7–G17. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; Van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Bonnet, M.; Buc, E.; Sauvanet, P.; Darcha, C.; Dubois, D.; Pereira, B.; Déchelotte, P.; Bonnet, R.; Pezet, D.; Darfeuille-Michaud, A. Colonization of the Human Gut by E. coli and Colorectal Cancer Risk. Clin. Cancer Res. 2014, 20, 859–867. [Google Scholar] [CrossRef] [Green Version]
- Martin, H.M.; Campbell, B.J.; Hart, C.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef]
- Wu, N.; Yang, X.; Zhang, R.; Li, J.; Xiao, X.; Hu, Y.; Chen, Y.; Yang, F.; Lu, N.; Wang, Z.; et al. Dysbiosis Signature of Fecal Microbiota in Colorectal Cancer Patients. Microb. Ecol. 2013, 66, 462–470. [Google Scholar] [CrossRef]
- Nakatsu, G.; Zhou, H.; Wu, W.K.; Wong, S.H.; Coker, O.O.; Dai, Z.; Li, X.; Szeto, C.H.; Sugimura, N.; Lam, T.Y.-T.; et al. Alterations in Enteric Virome Are Associated With Colorectal Cancer and Survival Outcomes. Gastroenterology 2018, 155, 529–541.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Metzger, R.N.; Krug, A.B.; Eisenächer, K. Enteric Virome Sensing—Its Role in Intestinal Homeostasis and Immunity. Viruses 2018, 10, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitetta, L.; Vitetta, G.; Hall, S. Immunological Tolerance and Function: Associations Between Intestinal Bacteria, Probiotics, Prebiotics, and Phages. Front. Immunol. 2018, 9, 2240. [Google Scholar] [CrossRef] [Green Version]
- Clooney, A.G.; Sutton, T.D.; Shkoporov, A.N.; Holohan, R.K.; Daly, K.M.; O’Regan, O.; Ryan, F.; Draper, L.A.; Plevy, S.E.; Ross, R.; et al. Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease. Cell Host Microbe 2019, 26, 764–778.e5. [Google Scholar] [CrossRef]
- Zuo, T.; Lu, X.-J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Coffey, M.J.; Low, I.; Stelzer-Braid, S.; Wemheuer, B.; Garg, M.; Thomas, T.; Jaffe, A.; Rawlinson, W.D.; Ooi, C.Y. The intestinal virome in children with cystic fibrosis differs from healthy controls. PLoS ONE 2020, 15, e0233557. [Google Scholar] [CrossRef]
- Gutierrez, M.W.; Arrieta, M.-C. The intestinal mycobiome as a determinant of host immune and metabolic health. Curr. Opin. Microbiol. 2021, 62, 8–13. [Google Scholar] [CrossRef]
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the healthy human gastrointestinal tract. Virulence 2016, 8, 352–358. [Google Scholar] [CrossRef]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef] [PubMed]
- Li, X.V.; Leonardi, I.; Iliev, I.D. Gut Mycobiota in Immunity and Inflammatory Disease. Immunity 2019, 50, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.-P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Limon, J.J.; Tang, J.; Li, D.; Wolf, A.J.; Michelsen, K.S.; Funari, V.; Gargus, M.; Nguyen, C.; Sharma, P.; Maymi, V.I.; et al. Malassezia Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Models. Cell Host Microbe 2019, 25, 377–388.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoarau, G.; Mukherjee, P.K.; Gower, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. mBio 2016, 7, e01250-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemoinne, S.; Kemgang, A.; Ben Belkacem, K.; Straube, M.; Jegou, S.; Corpechot, C.; Chazouillères, O.; Housset, C.; Sokol, H.; Network, S.-A.I. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut 2020, 69, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.-M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841. [Google Scholar] [CrossRef] [Green Version]
- Mar Rodríguez, M.; Pérez, D.; Javier Chaves, F.; Esteve, E.; Marin-Garcia, P.; Xifra, G.; Vendrell, J.; Jové, M.; Pamplona, R.; Ricart, W.; et al. Obesity changes the human gut mycobiome. Sci. Rep. 2015, 5, 14600. [Google Scholar] [CrossRef]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef]
- Hardouin, P.; Chiron, R.; Marchandin, H.; Armengaud, J.; Grenga, L. Metaproteomics to Decipher CF Host-Microbiota Interactions: Overview, Challenges and Future Perspectives. Genes 2021, 12, 892. [Google Scholar] [CrossRef]
- García-Durán, C.; Martínez-López, R.; Zapico, I.; Pérez, E.; Romeu, E.; Arroyo, J.; Hernáez, M.L.; Pitarch, A.; Monteoliva, L.; Gil, C. Distinct Human Gut Microbial Taxonomic Signatures Uncovered With Different Sample Processing and Microbial Cell Disruption Methods for Metaproteomic Analysis. Front. Microbiol. 2021, 12, 618566. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tam, R.Y.; van Dorst, J.M.; McKay, I.; Coffey, M.; Ooi, C.Y. Intestinal Inflammation and Alterations in the Gut Microbiota in Cystic Fibrosis: A Review of the Current Evidence, Pathophysiology and Future Directions. J. Clin. Med. 2022, 11, 649. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11030649
Tam RY, van Dorst JM, McKay I, Coffey M, Ooi CY. Intestinal Inflammation and Alterations in the Gut Microbiota in Cystic Fibrosis: A Review of the Current Evidence, Pathophysiology and Future Directions. Journal of Clinical Medicine. 2022; 11(3):649. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11030649
Chicago/Turabian StyleTam, Rachel Y., Josie M. van Dorst, Isabelle McKay, Michael Coffey, and Chee Y. Ooi. 2022. "Intestinal Inflammation and Alterations in the Gut Microbiota in Cystic Fibrosis: A Review of the Current Evidence, Pathophysiology and Future Directions" Journal of Clinical Medicine 11, no. 3: 649. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11030649