
The Evolutionary Scenario of Pediatric Unclassified Primary Antibody Deficiency to Adulthood
 , , , , ,
, , , , ,
Abstract
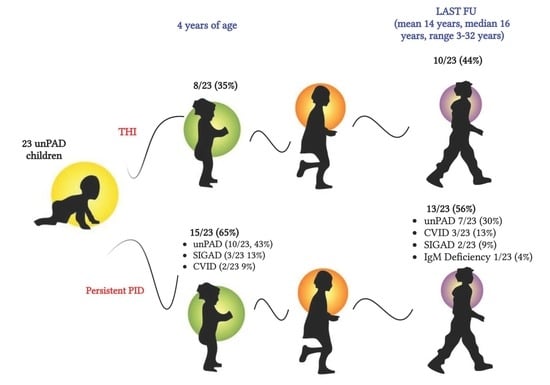
:
1. Introduction
2. Materials and Methods
3. Statistical Analysis
4. Results
4.1. Clinical and Immunological Findings at Initial Diagnosis, According to Age (<24 or >24 Months)
4.2. Clinical and Immunological Findings at 4 Years of Age and at Last FU (Mean 14 Years, Range 3–32 Years, Median 16 Years)
4.3. Clinical and Immunological Findings at Diagnosis of Patients with a Definitive Diagnosis of Persistent PID vs. THI
4.4. Genetic Characterization
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CVID | common variable immunodeficiency |
| ELISA | enzyme-linked immunosorbent assay |
| ESID | European Society for Immunodeficiencies |
| FACS | fluorescence-activated cell sorting |
| FU | follow-up |
| GI | gastrointestinal infections |
| IEI | inborn error of immunity |
| IgMD | IgM deficiency |
| IRT | immunoglobulin replacement therapy |
| LRTI | lower respiratory tract infection |
| NGS | next-generation sequencing |
| PAD | primary antibody deficiency |
| PID | primary immunodeficiency |
| PCP | pneumococcal |
| SIGAD | selective IgA deficiency |
| sIgE | serum IgE |
| SPT | skin prick tests |
| THI | transient hypogammaglobulinemia of infancy |
| TNFRSF13B/TACI | TNF receptor superfamily member 13B |
| TT | tetanus toxoid |
| UnPAD | unclassified primary antibody deficiency |
| UTI | urinary tract infections |
| URTI | upper respiratory tract infections |
References
- Vivarelli, E.; Matucci, A.; Bormioli, S.; Parronchi, P.; Liotta, F.; Cosmi, L.; Almerigogna, F.; Vultaggio, A. Effectiveness of low-dose intravenous immunoglobulin therapy in minor primary antibody deficiencies: A 2-year real-life experience. Clin. Exp. Immunol. 2021, 205, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Lougaris, V.; Pession, A.; Baronio, M.; Soresina, A.; Rondelli, R.; Gazzurelli, L.; Benvenuto, A.; Martino, S.; Gattorno, M.; Biondi, A.; et al. The Italian Registry for Primary Immunodeficiencies (Italian Primary Immunodeficiency Network; IPINet): Twenty Years of Experience (1999–2019). J. Clin. Immunol. 2020, 40, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, R.; Habibi, S.; Sharifi, L.; Azizi, G.; Abolhassani, H.; Olbrich, P.; Aghamohammadi, A. Common Variable Immunodeficiency: Epidemiology, Pathogenesis, Clinical Manifestations, Diagnosis, Classification, and Management. J. Investig. Allergol. Clin. Immunol. 2020, 30, 14–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filion, C.A.; Taylor-Black, S.; Sweater, P.J.; Radigan, L.; Cunningham-Rundles, C. Differentiation of Common Variable Immunodeficiency from IgG Deficiency. J. Allergy Clin. Immunol. Pract. 2018, 7, 1277–1284. [Google Scholar] [CrossRef]
- Keles, S.; Artac, H.; Kara, R.; Gokturk, B.; Ozen, A.; Reisli, I. Transient hypogammaglobulinemia and unclassified hypogammaglobulinemia: ‘Similarities and differences’. Pediatr. Allergy Immunol. 2010, 21, 843–851. [Google Scholar] [CrossRef]
- Kutukculer, N.; Gulez, N. The outcome of patients with unclassified hypogammaglobulinemia in early childhood. Pediatr. Allergy Immunol. 2009, 20, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Janssen, L.M.A.; Bassett, P.; Macken, T.; Van Esch, J.; Pruijt, H.; Knoops, A.; Sköld, M.; Parker, A.; DeVries, J.; de Vries, E. Mild Hypogammaglobulinemia Can Be a Serious Condition. Front. Immunol. 2018, 9, 2384. [Google Scholar] [CrossRef] [Green Version]
- Shehata, N.; Palda, V.; Bowen, T.; Haddad, E.; Issekutz, T.B.; Mazer, B.; Schellenberg, R.; Warrington, R.; Easton, D.; Anderson, D.; et al. The Use of Immunoglobulin Therapy for Patients With Primary Immune Deficiency: An Evidence-Based Practice Guideline. Transfus. Med. Rev. 2010, 24 (Suppl. 1), S28–S50. [Google Scholar] [CrossRef]
- Modell, V.; Gee, B.; Lewis, D.B.; Orange, J.S.; Roifman, C.M.; Routes, J.M.; Sorensen, R.U.; Notarangelo, L.D.; Modell, F. Global study of primary immunodeficiency diseases (PI)–diagnosis, treatment, and economic impact: An updated report from the Jeffrey Modell Foundation. Immunol. Res. 2011, 51, 61–70. [Google Scholar] [CrossRef]
- Wasserman, R. L Personalized Therapy: Immunoglobulin Replacement for Antibody Deficiency. Immunol. Allergy Clinic. North Am. 2019, 39, 95–111. [Google Scholar] [CrossRef]
- Khokar, A.; Gupta, S. Clinical and Immunological Features of 78 Adult Patients with Primary Selective IgG Subclass Deficiencies. Arch. Immunol. Et Ther. Exp. 2019, 67, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Karaman, S.; Erdem, S.B.; Gülez, N.; Genel, F. The Significance of B-cell Subsets in Patients with Unclassified Hypogammaglobulinemia and Association with Intravenous Immunoglobulin Replacement Requirement. Iran. J. Immunol. 2018, 15, 1–13. [Google Scholar] [PubMed]
- Cifaldi, C.; Bridget, I.; Barzaghi, F.; Zoccolillo, M.; Ferradini, V.; Petricone, D.; Cicalese, M.P.; Lazarevic, D.; Cittaro, D.; Omrani, M.; et al. Targeted NGS Platforms for Genetic Screening and Gene Discovery in Primary Immunodeficiencies. Front. Immunol. 2019, 10, 316. [Google Scholar] [CrossRef] [Green Version]
- Thalhammer, J.; Kindle, G.; Nieters, A.; Rusch, S.; Seppänen, M.R.; Fischer, A.; Grimbacher, B.; Edgar, D.; Buckland, M.; Mahlaoui, N.; et al. Initial presenting manifestations in 16,486 patients with inborn errors of immunity include infections and noninfectious manifestations. J. Allergy Clin. Immunol. 2021, 148, 1332–1341.e5. [Google Scholar] [CrossRef]
- Raje, N.; Dinakar, C. Overview of Immunodeficiency Disorders. Immunol. Allergy Clinic. North Am. 2015, 35, 599–623. [Google Scholar] [CrossRef] [Green Version]
- Modell, V.; Quinn, J.; Ginsberg, G.; Gladue, R.; Orange, J.; Modell, F. Modeling strategy to identify with primary immunodeficiency patients utilizing risk management and outcome measurement. Immunol. Res. 2017, 65, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, A.; Sayyahfar, S.; Soltani, Z.; Khodadost, M.; Moazzami, B.; Rezaei, N. Evaluation of the frequency and diagnostic delay of primary immunodeficiency disorders among suspected patients based on the 10 warning sign criteria: A cross-sectional study in Iran. Allergol. Immunopathol. 2020, 48, 711–719. [Google Scholar] [CrossRef]
- Slade, C.A.; Bosco, J.J.; Giang, T.B.; Kruse, E.; Stirling, R.G.; Cameron, P.U.; Hore-Lacy, F.; Sutherland, M.F.; Barnes, S.L.; Holdsworth, S.; et al. Delayed Diagnosis and Complications of Predominantly Antibody Deficiencies in a Cohort of Australian Adults. Front. Immunol. 2018, 9, 694. [Google Scholar] [CrossRef] [Green Version]
- Janssen, L.M.A.; Reijnen, I.C.G.M.; Milito, C.; Edgar, D.; Chapel, H.; de Vries, E. The unPAD consortium Protocol for the unclassified primary antibody deficiency (unPAD) study: Characterization and classification of patients using the ESID online Registry. PLoS ONE 2022, 17, e0266083. [Google Scholar] [CrossRef]
- Pickett, G.; Motazedi, T.; Kutac, C.; Cahill, G.; Cunnigham-Rundles, C.; Fuleihan, R.L.; Sullivan, K.E.; Rider, N.L. Infection Phenotypes Among Patients with Primary Antibody Deficiency Mined from a US Patient Registry. J. Clin. Immunol. 2021, 41, 374–381. [Google Scholar] [CrossRef]
- Demirdag, Y.Y.; Gupta, S. Update on Infections in Primary Antibody Deficiencies. Front. Immunol. 2021, 12, 634181. [Google Scholar] [CrossRef] [PubMed]
- Moschese, V.; Graziani, S.; Avanzini, M.; Carsetti, R.; Marconi, M.; LaRocca, M.; Chini, L.; Pignata, C.; Soresina, A.; Consolini, R.; et al. A Prospective Study on Children with Initial Diagnosis of Transient Hypogammaglobulinemia of Infancy: Results from the Italian Primary Immunodeficiency Network. Int. J. Immunopathol. Pharmacol. 2008, 21, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameratunga, R.; Ahn, Y.; Steele, R.; Woon, S. Transient hypogammaglobulinaemia of infancy: Many patients recover in adolescence and adulthood. Clin. Exp. Immunol. 2019, 198, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Seijas, A.B.; Graziani, S.; Cancrini, C.; Finocchi, A.; Ferrari, S.; Miniero, R.; Conti, F.; Zuntini, R.; Chini, L.; Chiarello, P.; et al. The Impact of TACI Mutations: From Hypogammaglobulinemia in Infancy to Autoimmunity in Adulthood. Int. J. Immunopathol. Pharmacol. 2012, 25, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Salzer, U.; Bacchelli, C.; Buckridge, S.; Pan-Hammarström, Q.; Jennings, S.; Lougaris, V.; Bergbreiter, A.; Hagena, T.; Birmelin, J.; Plebani, A.; et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood 2009, 113, 1967–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eroglu, F.K.; Kaya, F.A.; Cagdas, D.; Özgur, T.T.; Yilmaz, T.; Tezcan, I.; Sanal, O. B lymphocyte subsets and outcomes in patients with an initial diagnosis of transient hypogammaglobulinemia of infancy. SCAN J. Immunol. 2018, 88, e12709. [Google Scholar] [CrossRef]
- Moschese, V.; Carsetti, R.; Graziani, S.; Chini, L.; Soresina, A.R.; LaRocca, M.; Bossi, G.; DiCesare, S.; Plebani, A.; Italian Primary Immunodeficiency Network. Memory B-cell subsets as a predictive marker of outcome in hypogammaglobulinemia during infancy. J. Allergy Clin. Immunol. 2007, 120, 474–476. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| unPAD | <24 Months (10 pts) | >24 Months (13 pts) | p-Value | |
|---|---|---|---|---|
| CLINICAL MANIFESTATIONS | 22/23 (96%) | 10/10 (100%) | 12/13 (92%) | ns |
| Infections | 22/22(100%) | 9/10 (90%) | 12/12 (100%) | ns |
| URTI | 12/22 (55%) | 6/10 (60%) | 6/12 (50%) | ns |
| LRTI | 10/22 (45%) | 4/10 (40%) | 6/12 (50%) | ns |
| GI | 5/22 (23%) | 1/10 (10%) | 4/12 (33%) | ns |
| UTI | 5/22(23%) | 4/10 (40%) | 1/12 (8%) | ns |
| FEVER | 1/22 (5%) | 1/10 (10%) | 0/12 (0%) | ns |
| SKIN | 1/22 (5%) | 1/10 (10%) | 0/12 (0%) | ns |
| Allergy | 7/22 (32%) | 3/10 (30%) | 4/12 (33%) | ns |
| Asthma | 2/7 (29%) | 0/3 (0%) | 2/4 (50%) | ns |
| Atopic dermatitis | 3/7 (43%) | 2/3 (67%) | 1/4 (25%) | ns |
| Food allergy | 2/7 (29%) | 1/3 (33%) | 1/4 (25%) | ns |
| Conjiunctivitis | 1/7 (14%) | 1/3 (33%) | 0/4 (0%) | ns |
| Rhinitis | 4/7 (57%) | 1/3 (33%) | 3/4 (75%) | ns |
| Autoimmunity | 2/22 (9%) | 2/10 (20%) | 0/12 (0%) | ns |
| Neutropenia | 2/2 (100%) | 2/2 (100%) | - | ns |
| IMMUNOLOGICAL ABNORMALITIES | 23/23 (100%) | 10/10 (100%) | 13/13 (100%) | ns |
| Isolated or combined IgG defect | 19/23 (82%) | 9/10 (90%) | 10/13 (77%) | ns |
| Isolated or combined IgA defect | 14/23 (61%) | 4/10 (40%) | 8/13 (62%) | ns |
| Isolated or combined IgM defect | 9/23 (39%) | 1/10 (10%) | 8/13 (62%) | 0.028 |
| Combined IgG defect | 13/23 (56%) | 6/10 (60%) | 7/13 (54%) | ns |
| Combined IgA defect | 13/23 (56%) | 5/10 (50%) | 8/13 (62%) | ns |
| Combined IgM defect | 8/23 (35%) | 0/10 (0%) | 8/13 (62%) | 0.0075 |
| IgG subclasses defect | 7/9 (77%) | 1/2 (50%) | 6/7 (86%) | ns |
| Low anti TT antibody response | 4/18 (22%) | 3/7 (43%) | 1/11 (9%) | ns |
| Low anti PCP antibody response | 7/19 (37%) | 2/7 (29%) | 5/12 (16%) | ns |
| Low switched memory B cells | 10/18 (55%) | 5/7 (71%) | 5/11 (45%) | ns |
| Low IgM memory B cells | 1/18 (5%) | 1/7 (14%) | 0/11 (0%) | ns |
| Persistent PIDs (13 pts) | THI (10 pts) | p-Value | |
|---|---|---|---|
| CLINICAL MANIFESTATIONS | |||
| Infections | 5/13 (38%) | 0/10 (0%) | 0.04 |
| URTI | 3/5 (60%) | - | 0.02 |
| LRTI | 3/5 (60%) | - | 0.02 |
| GI | 1/5 (20%) | - | ns |
| Allergy | 6/13 (46%) | 5/10 (50%) | ns |
| Asthma | 2/6 (33%) | 0/5 (0%) | ns |
| Rhinitis | 3/6 (50%) | 4/5 (80%) | ns |
| Conjiunctivitis | 1/6 (17%) | 2/5 (40%) | ns |
| Autoimmunity | 2/13 (15%) | 0/10 (0%) | ns |
| Neutropenia | 2/2 (100%) | - | ns |
| Vasculitis | 1/2 (50%) | - | ns |
| IMMUNOLOGICAL ABNORMALITIES | 13/13 (100%) | 0/10 (0%) | 0.0001 |
| Isolated or combined IgG defect | 6/13 (46%) | 0/10 (0%) | 0.02 |
| Isolated or combined IgA defect | 8/13 (61%) | 0/10 (0%) | 0.003 |
| Isolated or combined IgM defect | 7/13 (54%) | 0/10 (0%) | 0.007 |
| IgG subclass defect | 7/13 (54%) | 0/10 (0%) | 0.007 |
| Low anti TT antibody response | 1/9 (11%) | 0/10 (0%) | ns |
| Low anti PCP antibody response | 2/10 (20%) | 0/10 (0%) | ns |
| Low switched memory B cells | 7/13 (54%) | 0/10 (0%) | ns |
| Low IgM memory B cells | 2/13 (15%) | 0/10 (0%) | ns |
| Genetic Characterization | 8/13 (61%) | - | |
| TNFRSF13B mutations | 4/8 (50%) | ||
| TNFRSF13B c.301T>C plus TNFRSF13B c.204dupA | 2/4 (50%) | ||
| Heterozygous TNFRSF13B c.301T>C mutation | 2/4 (50%) |
| Persistent PIDs (13 pts) | THI (10 pts) | Univariate Analysis p-Value | Logistic Regression p-Value | |
|---|---|---|---|---|
| Positive Family History for PID | 5/13 (38%) | 1/10 (10%) | ns | |
| CLINICAL MANIFESTATIONS | ||||
| Infections | 12/13 (92%) | 10/10 (100%) | ns | |
| LRTI | 8/12 (67%) | 2/10 (20%) | 0.04 | <0.05 |
| Allergy | 4/13 (31%) | 3/10 (30%) | ns | |
| Autoimmunity | 1/13 (8%) | 1/10 (10%) | ns | |
| IMMUNOLOGICAL ABNORMALITIES | ||||
| Isolated or combined IgG defect | 11/13 (85%) | 8/10 (80%) | ns | |
| Isolated or combined IgA defect | 11/13 (85%) | 3/10 (30%) | 0.0013 | <0.05 |
| Isolated or combined IgM defect | 6/13 (46%) | 3/10 (30%) | ns | |
| IgG subclass defect | 5/5 (100%) | 2/4 (50%) | ns | |
| Low anti PCP antibody response | 7/11 (64%) | 0/8 (0%) | 0.0128 | <0.05 |
| Low switched memory B cells | 7/13 (54%) | 0/5 (0%) | ns | |
| Low IgM memory B cells | 1/13 (8%) | 0/5(0%) | ns |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sgrulletti, M.; Costagliola, G.; Giardino, G.; Graziani, S.; Del Duca, E.; Di Cesare, S.; Di Matteo, G.; Consolini, R.; Pignata, C.; Moschese, V. The Evolutionary Scenario of Pediatric Unclassified Primary Antibody Deficiency to Adulthood. J. Clin. Med. 2023, 12, 4206. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm12134206
Sgrulletti M, Costagliola G, Giardino G, Graziani S, Del Duca E, Di Cesare S, Di Matteo G, Consolini R, Pignata C, Moschese V. The Evolutionary Scenario of Pediatric Unclassified Primary Antibody Deficiency to Adulthood. Journal of Clinical Medicine. 2023; 12(13):4206. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm12134206
Chicago/Turabian StyleSgrulletti, Mayla, Giorgio Costagliola, Giuliana Giardino, Simona Graziani, Elisabetta Del Duca, Silvia Di Cesare, Gigliola Di Matteo, Rita Consolini, Claudio Pignata, and Viviana Moschese. 2023. "The Evolutionary Scenario of Pediatric Unclassified Primary Antibody Deficiency to Adulthood" Journal of Clinical Medicine 12, no. 13: 4206. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm12134206