Reformulating Pro-Oxidant Microglia in Neurodegeneration

 , , ,
, , ,
Abstract
:1. Microglia: Origin and Roles in Health and Disease; Triggering Receptors Expressed on Myeloid Cells-2 (TREM2) Significance
2. Sensing the Disease-Associated Environment: TLRs and TREM2, Main Drivers of Microglia Polarization
2.1. Toll-Like Receptors (TLRs)
2.2. TREM2
3. Microglia Subtypes Under Conditions of Neurodegeneration
3.1. Classical M1-Like and M2-Like Phenotypes
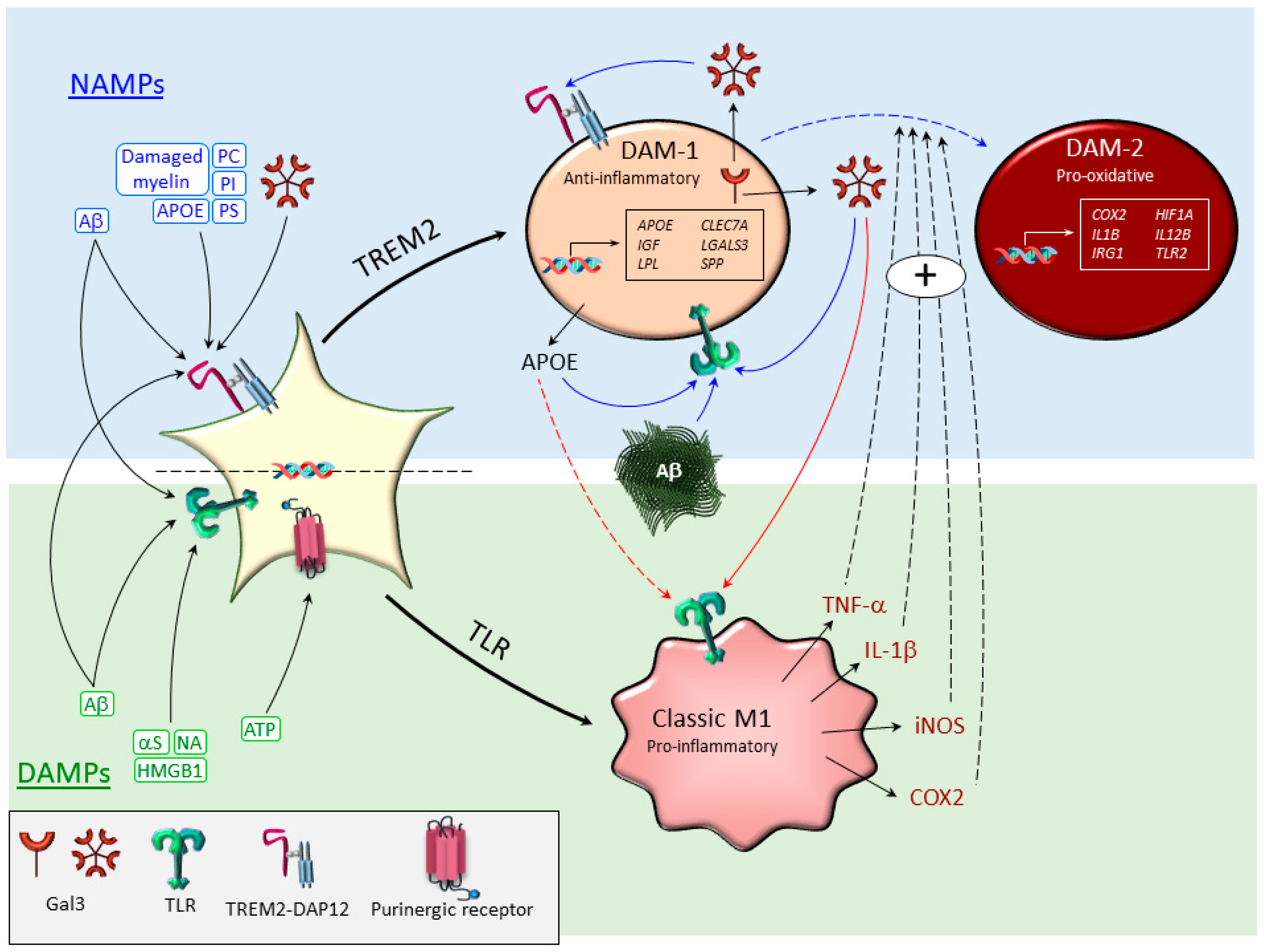
3.2. Disease-Associated Microglia (DAM)
3.3. Necroptotic Microglia
3.4. Dark Microglia
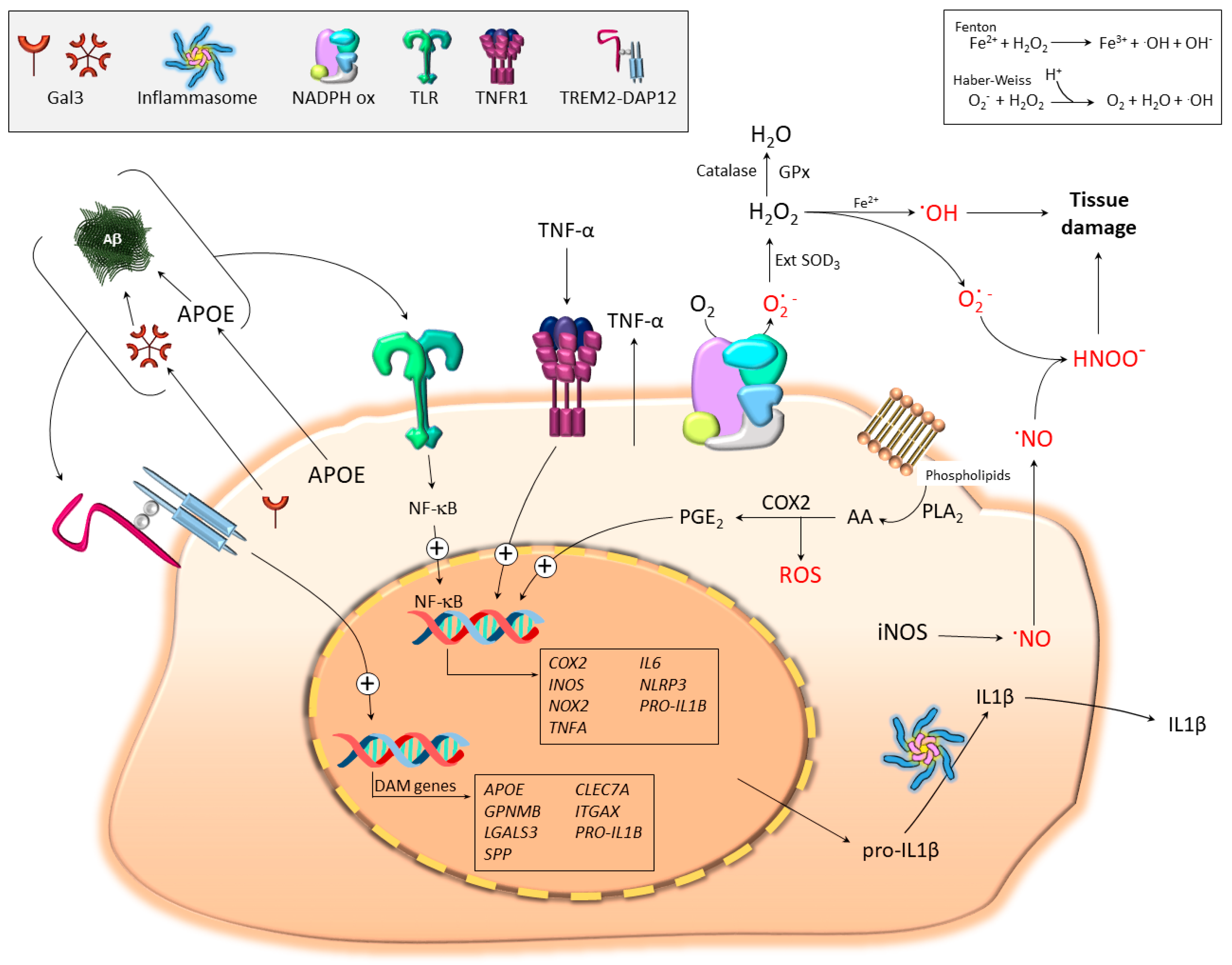
4. Microglial Contribution to Oxidative Stress
4.1. The Basis: Free Radical Generation in Biological Systems
4.2. Pro-Oxidant Microglia Profiling: iNOS, COX2, TNF-α, and IL-1β
4.2.1. iNOS and Nitric Oxide (NO)
4.2.2. Cyclooxygenase-2 (COX2)
4.2.3. Tumor Necrosis Factor α (TNF-α)
4.2.4. Interleukin 1β (IL-1β)
5. Role of Oxidative Stress in Pathological Protein Aggregates in Alzheimer’s and Parkinson’s Diseases
6. Epigenetic Regulation of Microglia Under Oxidative Stress Conditions in Neurodegeneration
7. Clinical Studies Using Anti-Inflammatory/Antioxidative Compounds
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
References
- Del Rio-Hortega, P. El tercer elemento de los centros nerviosos. Bol. Soc. Esp. Biol. 1919, 9, 69–120. [Google Scholar]
- Del Rio-Hortega, P. The microglia. Lancet 1939, 1, 1023–1026. [Google Scholar] [CrossRef]
- Alliot, F.; Godin, I.; Pessac, B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Dev. Brain Res. 1999, 117, 145–152. [Google Scholar] [CrossRef]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.A.; Boddeke, H.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Shen, X.; Venero, J.L.; Joseph, B.; Burguillos, M.A. Caspases orchestrate microglia instrumental functions. Prog. Neurobiol. 2018, 171, 50–71. [Google Scholar] [CrossRef]
- Verheijen, J.; Sleegers, K. Understanding Alzheimer Disease at the Interface between Genetics and Transcriptomics. Trends Genet. 2018, 34, 434–447. [Google Scholar] [CrossRef] [Green Version]
- Hammond, T.R.; Marsh, S.E.; Stevens, B. Immune Signaling in Neurodegeneration. Immunity 2019, 50, 955–974. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Hamerman, J.A.; Ni, M.; Killebrew, J.R.; Chu, C.L.; Lowell, C.A. The expanding roles of ITAM adapters FcRgamma and DAP12 in myeloid cells. Immunol. Rev. 2009, 232, 42–58. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Toll-like receptors in the pathogenesis of neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; De Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Lehnardt, S. Innate immunity and neuroinflammation in the CNS: The role of microglia in Toll-like receptor-mediated neuronal injury. Glia 2010, 58, 253–263. [Google Scholar] [CrossRef]
- Okun, E.; Griffioen, K.J.; Mattson, M.P. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011, 34, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar amyloid-beta peptides activate microglia via TLR2: Implications for Alzheimer’s disease. J. Immunol. 2008, 181, 7254–7262. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Lee, S.J.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Agalave, N.M.; Larsson, M.; Abdelmoaty, S.; Su, J.; Baharpoor, A.; Lundbäck, P.; Palmblad, K.; Andersson, U.; Harris, H.; Svensson, C.I. Spinal HMGB1 induces TLR4-mediated long-lasting hypersensitivity and glial activation and regulates pain-like behavior in experimental arthritis. Pain 2014, 155, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Burguillos, M.A.; Svensson, M.; Schulte, T.; Boza-Serrano, A.; Garcia-Quintanilla, A.; Kavanagh, E.; Santiago, M.; Viceconte, N.; Oliva-Martin, M.J.; Osman, A.M.; et al. Microglia-Secreted Galectin-3 Acts as a Toll-like Receptor 4 Ligand and Contributes to Microglial Activation. Cell Rep. 2015, 10, 1626–1638. [Google Scholar] [CrossRef] [Green Version]
- Yip, P.K.; Carrillo-Jimenez, A.; King, P.; Vilalta, A.; Nomura, K.; Chau, C.C.; Egerton, A.M.S.; Liu, Z.H.; Shetty, A.J.; Tremoleda, J.L.; et al. Galectin-3 released in response to traumatic brain injury acts as an alarmin orchestrating brain immune response and promoting neurodegeneration. Sci. Rep. 2017, 7, 41689. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- DeLeo, F.R.; Renee, J.; McCormick, S.; Nakamura, M.; Apicella, M.; Weiss, J.P.; Nauseef, W.M. Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J. Clin. Investig. 1998, 101, 455–463. [Google Scholar] [CrossRef]
- El-Benna, J.; Dang, P.M.C.; Gougerot-Pocidalo, M.A.; Elbim, C. Phagocyte NADPH oxidase: A multicomponent enzyme essential for host defenses. Arch. Immunol. Ther. Exp. 2005, 53, 199–206. [Google Scholar]
- Qian, L.; Wei, S.J.; Zhang, D.; Hu, X.; Xu, Z.; Wilson, B.; El-Benna, J.; Hong, J.S.; Flood, P.M. Potent anti-inflammatory and neuroprotective effects of TGF-beta1 are mediated through the inhibition of ERK and p47phox-Ser345 phosphorylation and translocation in microglia. J. Immunol. 2008, 181, 660–668. [Google Scholar] [CrossRef]
- Shimohama, S.; Tanino, H.; Kawakami, N.; Okamura, N.; Kodama, H.; Yamaguchi, T.; Hayakawa, T.; Nunomura, A.; Chiba, S.; Perry, G.; et al. Activation of NADPH Oxidase in Alzheimer’s Disease Brains. Biochem. Biophys. Res. Commun. 2000, 273, 5–9. [Google Scholar] [CrossRef]
- Wu, D.C.; Teismann, P.; Tieu, K.; Vila, M.; Jackson-Lewis, V.; Ischiropoulos, H.; Przedborski, S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 6145–6150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Qin, L.; Gao, H.M.; Wilson, B.; Ali, S.F.; Hong, J.S.; Liu, B. Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: Role of NADPH oxidase. FASEB J. 2004, 18, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Valencia, A.; Sapp, E.; Kimm, J.S.; McClory, H.; Reeves, P.B.; Alexander, J.; Ansong, K.A.; Masso, N.; Frosch, M.P.; Kegel, K.B.; et al. Elevated NADPH oxidase activity contributes to oxidative stress and cell death in Huntington’s disease. Hum. Mol. Genet. 2013, 22, 1112–1131. [Google Scholar] [CrossRef]
- Chéret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.H.; Mallat, M. Neurotoxic Activation of Microglia Is Promoted by a Nox1-Dependent NADPH Oxidase. J. Neurosci. 2008, 28, 12039–12051. [Google Scholar] [CrossRef]
- Miletic, A.V.; Graham, D.B.; Montgrain, V.; Fujikawa, K.; Kloeppel, T.; Brim, K.; Weaver, B.; Schreiber, R.; Xavier, R.; Swat, W. Vav proteins control MyD88-dependent oxidative burst. Blood 2006, 109, 3360–3368. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Kim, H.P.; Geng, X.H.; Nakao, A.; Wang, X.; Murase, N.; Drain, P.F.; Wang, X.; Sasidhar, M.; Nabel, E.G.; et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J. Exp. Med. 2006, 203, 2377–2389. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.W.; Kwon, M.J.; Choi, A.M.K.; Kim, H.P.; Nakahira, K.; Hwang, D.H. Fatty Acids Modulate Toll-like Receptor 4 Activation through Regulation of Receptor Dimerization and Recruitment into Lipid Rafts in a Reactive Oxygen Species-dependent Manner. J. Biol. Chem. 2009, 284, 27384–27392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzawa, A.; Saegusa, K.; Noguchi, T.; Sadamitsu, C.; Nishitoh, H.; Nagai, S.; Koyasu, S.; Matsumoto, K.; Takeda, K.; Ichijo, H. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 2005, 6, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Lao, Y.; Yang, K.; Wang, Z.; Sun, X.; Zou, Q.; Yu, X.; Cheng, J.; Tong, X.; Yeh, E.T.H.; Yang, J.; et al. DeSUMOylation of MKK7 kinase by the SUMO2/3 protease SENP3 potentiates lipopolysaccharide-induced inflammatory signaling in macrophages. J. Biol. Chem. 2018, 293, 3965–3980. [Google Scholar] [CrossRef] [Green Version]
- Cannon, J.P.; O’Driscoll, M.; Litman, G.W. Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics 2012, 64, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; Chen, T.T.; Tchao, N.K.; Seaman, W.E. Pattern recognition by TREM-2: Binding of anionic ligands. J. Immunol. 2003, 171, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Tan, C.C.; Hou, X.H.; Cao, X.P.; Tan, L.; Yu, J.T. TREM2 Variants and Neurodegenerative Diseases: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2019, 68, 1171–1184. [Google Scholar] [CrossRef]
- Jay, T.R.; Von Saucken, V.E.; Landreth, G.E. TREM2 in Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 56. [Google Scholar] [CrossRef]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; Van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Jordão, M.J.C.; Sankowski, R.; Brendecke, S.M.; Sagar; Locatelli, G.; Tai, Y.H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363. [Google Scholar] [CrossRef]
- Matarin, M.; Salih, D.A.; Yasvoina, M.; Cummings, D.M.; Guelfi, S.; Liu, W.; Solim, M.A.N.; Moens, T.G.; Paublete, R.M.; Ali, S.S.; et al. A Genome-wide Gene-Expression Analysis and Database in Transgenic Mice during Development of Amyloid or Tau Pathology. Cell Rep. 2015, 10, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Serrano, J.R.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Ripoll, V.M.; Irvine, K.M.; Ravasi, T.; Sweet, M.J.; Hume, D.A. Gpnmb Is Induced in Macrophages by IFN-γ and Lipopolysaccharide and Acts as a Feedback Regulator of Proinflammatory Responses. J. Immunol. 2007, 178, 6557–6566. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Alboslemy, T.; Safadi, F.; Kim, M.H. Glycoprotein Nonmelanoma Clone B Regulates the Crosstalk between Macrophages and Mesenchymal Stem Cells toward Wound Repair. J. Investig. Dermatol. 2017, 138, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhuo, H.; Ouyang, H.; Liu, Y.; Yuan, F.; Sun, L.; Liu, F.; Liu, H. Glycoprotein non-metastatic melanoma protein b (Gpnmb) is highly expressed in macrophages of acute injured kidney and promotes M2 macrophages polarization. Cell. Immunol. 2017, 316, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Budge, K.M.; Neal, M.L.; Richardson, J.R.; Safadi, F.F. Glycoprotein NMB: An Emerging Role in Neurodegenerative Disease. Mol. Neurobiol. 2017, 55, 5167–5176. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Castano, A.P.; Hudson, T.E.; Nowlin, B.T.; Lin, S.L.; Bonventre, J.V.; Swanson, K.D.; Duffield, J.S. The melanoma-associated transmembrane glycoprotein Gpnmb controls trafficking of cellular debris for degradation and is essential for tissue repair. FASEB J. 2010, 24, 4767–4781. [Google Scholar] [CrossRef] [PubMed]
- Metz, R.L.; Patel, P.S.; Hameed, M.; Bryan, M.; Rameshwar, P. Role of human HGFIN/nmb in breast cancer. Breast Cancer Res. 2007, 9, R58. [Google Scholar] [CrossRef]
- Zhong, L.; Chen, X.F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 92, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 2019, 22, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Vaccari, J.P.D.R.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031. [Google Scholar] [CrossRef]
- Zhong, L.; Wang, Z.; Wang, D.; Martens, Y.A.; Wu, L.; Xu, Y.; Wang, K.; Li, J.; Huang, R.; Can, D.; et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 2018, 13, 15. [Google Scholar] [CrossRef]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Poliani, P.L.; Wang, Y.; Fontana, E.; Robinette, M.L.; Yamanishi, Y.; Gilfillan, S.; Colonna, M. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Investig. 2015, 125, 2161–2170. [Google Scholar] [CrossRef] [Green Version]
- Boza-Serrano, A.; Ruiz, R.; Sanchez-Varo, R.; García-Revilla, J.; Yang, Y.; Jimenez-Ferrer, I.; Paulus, A.; Wennström, M.; Vilalta, A.; Allendorf, D.; et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 2019. [Google Scholar] [CrossRef]
- Lalancette-Hebert, M.; Swarup, V.; Beaulieu, J.M.; Bohacek, I.; Abdelhamid, E.; Weng, Y.C.; Sato, S.; Kriz, J. Galectin-3 Is Required for Resident Microglia Activation and Proliferation in Response to Ischemic Injury. J. Neurosci. 2012, 32, 10383–10395. [Google Scholar] [CrossRef] [Green Version]
- Gale, S.C.; Gao, L.; Mikacenic, C.; Coyle, S.M.; Rafaels, N.; Dudenkov, T.M.; Madenspacher, J.H.; Draper, D.W.; Ge, W.; Aloor, J.J.; et al. APOε4 is associated with enhanced in vivo innate immune responses in human subjects. J. Allergy Clin. Immunol. 2014, 134, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Doens, D.; Fernandez, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 Mediates the Innate Host Response to β-Amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef]
- Landel, V.; Baranger, K.; Virard, I.; Loriod, B.; Khrestchatisky, M.; Rivera, S.; Benech, P.; Féron, F. Temporal gene profiling of the 5XFAD transgenic mouse model highlights the importance of microglial activation in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 33. [Google Scholar] [CrossRef]
- Linnartz-Gerlach, B.; Bodea, L.G.; Klaus, C.; Ginolhac, A.; Halder, R.; Sinkkonen, L.; Walter, J.; Colonna, M.; Neumann, H. TREM2 triggers microglial density and age-related neuronal loss. Glia 2019, 67, 539–550. [Google Scholar] [CrossRef]
- Klesney-Tait, J.; Turnbull, I.R.; Colonna, M. The TREM receptor family and signal integration. Nat. Immunol. 2006, 7, 1266–1273. [Google Scholar] [CrossRef]
- Painter, M.M.; Atagi, Y.; Liu, C.C.; Rademakers, R.; Xu, H.; Fryer, J.D.; Bu, G. TREM2 in CNS homeostasis and neurodegenerative disease. Mol. Neurodegener. 2015, 10, 43. [Google Scholar] [CrossRef] [Green Version]
- Li, J.T.; Zhang, Y. TREM2 regulates innate immunity in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 107. [Google Scholar] [CrossRef]
- Silke, J.; Rickard, J.A.; Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 2015, 16, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2016, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Najjar, M.; Saleh, D.; Zelic, M.; Nogusa, S.; Shah, S.; Tai, A.; Finger, J.N.; Polykratis, A.; Gough, P.J.; Bertin, J.; et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor. Immunity 2016, 45, 46–59. [Google Scholar] [CrossRef]
- Buchrieser, J.; Oliva-Martin, M.J.; Moore, M.D.; Long, J.C.D.; Cowley, S.A.; Perez-Simón, J.A.; James, W.; Venero, J.L. RIPK1 is a critical modulator of both tonic and TLR-responsive inflammatory and cell death pathways in human macrophage differentiation. Cell Death Dis. 2018, 9, 973. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Vilalta, A.; Tolkovsky, A.M.; Brown, G.C. Caspase inhibitors protect neurons by enabling selective necroptosis of inflamed microglia. J. Biol. Chem. 2013, 288, 9145–9152. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.F.; Davies, C.L.; Holloway, R.K.; Labrak, Y.; Ireland, G.; Carradori, D.; Dillenburg, A.; Borger, E.; Soong, D.; Richardson, J.C.; et al. Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci. 2019, 22, 1046–1052. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef]
- Yang, S.H.; Lee, D.; Shin, J.; Lee, S.; Baek, S.; Kim, Y. Nec-1 Alleviates Cognitive Impairment with Reduction of Aβ and Tau Abnormalities in App/Ps1 Mice. EMBO Mol. Med. 2017, 9, 61–77. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sánchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [Green Version]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.; Joseph, B. Microglial subtypes: diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Lassègue, B.; Martín, A.S.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Tanaka, T.; Yoshida, Y.; Inagi, R. Physiological and pathophysiological role of reactive oxygen species and reactive nitrogen species in the kidney. Clin. Exp. Pharmacol. Physiol. 2018, 45, 1097–1105. [Google Scholar] [CrossRef]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [Green Version]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous non-enzymatic antioxidants in the human body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef]
- Cherubini, A.; Ruggiero, C.; Morand, C.; Lattanzio, F.; Dell’aquila, G.; Zuliani, G.; Di Iorio, A.; Andres-Lacueva, C. Dietary antioxidants as potential pharmacological agents for ischemic stroke. Curr. Med. Chem. 2008, 15, 1236–1248. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Zhu, X.; Smith, M.A.; Perry, G.; Aliev, G. Mitochondrial failures in Alzheimer’s disease. Am. J. Alzheimer’s Dis. Dement. 2004, 19, 345–352. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Carri, M.T.; Ferri, A.; Cozzolino, M.; Calabrese, L.; Rotilio, G. Neurodegeneration in amyotrophic lateral sclerosis: The role of oxidative stress and altered homeostasis of metals. Brain Res. Bull. 2003, 61, 365–374. [Google Scholar] [CrossRef]
- Giordano, G.; White, C.C.; McConnachie, L.A.; Fernandez, C.; Kavanagh, T.J.; Costa, L.G. Neurotoxicity of Domoic Acid in Cerebellar Granule Neurons in a Genetic Model of Glutathione Deficiency. Mol. Pharmacol. 2006, 70, 2116–2126. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Sahley, C.L.; Muller, K.J. ATP and NO dually control migration of microglia to nerve lesions. Dev. Neurobiol. 2009, 69, 60–72. [Google Scholar] [CrossRef]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Freudenberg, F.; Alttoa, A.; Reif, A. Neuronal nitric oxide synthase (NOS1) and its adaptor, NOS1AP, as a genetic risk factors for psychiatric disorders. Genes Brain Behav. 2015, 14, 46–63. [Google Scholar] [CrossRef]
- Necchi, D.; Virgili, M.; Monti, B.; Contestabile, A.; Scherini, E. Regional alterations of the NO/NOS system in the aging brain: A biochemical, histochemical and immunochemical study in the rat. Brain Res. 2002, 933, 31–41. [Google Scholar] [CrossRef]
- Min, K.J.; Pyo, H.K.; Yang, M.S.; Ji, K.A.; Jou, I.; Joe, E.H. Gangliosides activate microglia via protein kinase C and NADPH oxidase. Glia 2004, 48, 197–206. [Google Scholar] [CrossRef]
- Bal-Price, A.; Matthias, A.; Brown, G.C. Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J. Neurochem. 2002, 80, 73–80. [Google Scholar] [CrossRef]
- Mander, P.; Brown, G.C. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J. Neuroinflamm. 2005, 2, 20. [Google Scholar] [CrossRef]
- Trujillo, M.; Ferrer-Sueta, G.; Radi, R. Peroxynitrite Detoxification and Its Biologic Implications. Antioxid. Redox Signal. 2008, 10, 1607–1620. [Google Scholar] [CrossRef]
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Parthanatos, a messenger of death. Front. Biosci. 2009, 14, 1116–1128. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, K.W.; Kim, M.S.; Lee, H.J. Piceatannol attenuates hydrogen-peroxide-and peroxynitrite-induced apoptosis of PC12 cells by blocking down-regulation of Bcl-XL and activation of JNK. J. Nutr. Biochem. 2007, 19, 459–466. [Google Scholar] [CrossRef]
- Franco, M.C.; Ricart, K.C.; Gonzalez, A.S.; Dennys, C.N.; Nelson, P.A.; Janes, M.S.; Mehl, R.A.; Landar, A.; Estevez, A.G. Nitration of Hsp90 on Tyrosine 33 Regulates Mitochondrial Metabolism. J. Biol. Chem. 2015, 290, 19055–19066. [Google Scholar] [CrossRef] [Green Version]
- Doyle, K.M.; Kennedy, D.; Gorman, A.M.; Gupta, S.; Healy, S.J.M.; Samali, A. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med. 2011, 15, 2025–2039. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, R.G.; Ciznadija, D.; Vanevski, M.; Mantamadiotis, T. Transcriptional regulation of cyclo-oxygenase expression: Three pillars of control. Int. J. Immunopathol. Pharmacol. 2003, 16, 59–67. [Google Scholar]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in Inflammatory and Degenerative Brain Diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Teismann, P.; Vila, M.; Choi, D.K.; Tieu, K.; Wu, D.C.; Jackson-Lewis, V.; Przedborski, S. COX-2 and neurodegeneration in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 272–277. [Google Scholar] [CrossRef]
- Teismann, P. COX-2 in the neurodegenerative process of Parkinson’s disease. BioFactors 2012, 38, 395–397. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Perry, R.T.; Collins, J.S.; Harrell, L.E.; Acton, R.T.; Go, R.C. Investigation of association of 13 polymorphisms in eight genes in southeastern African American Alzheimer disease patients as compared to age-matched controls. Am. J. Med. Genet. 2001, 105, 332–342. [Google Scholar] [CrossRef]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-B is increased in dopaminergic neurons of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxid. Med. Cell Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef]
- Mander, P.K.; Jekabsone, A.; Brown, G.C. Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J. Immunol. 2006, 176, 1046–1052. [Google Scholar] [CrossRef]
- Jekabsone, A.; Mander, P.K.; Tickler, A.; Sharpe, M.; Brown, G.C. Fibrillar beta-amyloid peptide Aβ1–40 activates microglial proliferation via stimulating TNF-α release and H2O2 derived from NADPH oxidase: A cell culture study. J. Neuroinflamm. 2006, 3, 24. [Google Scholar] [CrossRef]
- Pawate, S.; Shen, Q.; Fan, F.; Bhat, N.R. Redox regulation of glial inflammatory response to lipopolysaccharide and interferon? J. Neurosci. Res. 2004, 77, 540–551. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- López-Castejón, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Brož, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2010, 469, 221–225. [Google Scholar] [CrossRef]
- Parajuli, B.; Sonobe, Y.; Horiuchi, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Oligomeric amyloid β induces IL-1β processing via production of ROS: Implication in Alzheimer’s disease. Cell Death Dis. 2013, 4, e975. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinson’s Dis. 2017, 3, 30. [Google Scholar] [CrossRef]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Bellezza, I.; Grottelli, S.; Costanzi, E.; Scarpelli, P.; Pigna, E.; Morozzi, G.; Mezzasoma, L.; Peirce, M.J.; Moresi, V.; Adamo, S.; et al. Peroxynitrite Activates the NLRP3 Inflammasome Cascade in SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2017, 55, 2350–2361. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef]
- Espay, A.J.; Vizcarra, J.A.; Marsili, L.; Lang, A.E.; Simon, D.K.; Merola, A.; Josephs, K.A.; Fasano, A.; Morgante, F.; Savica, R.; et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 2019, 92, 329–337. [Google Scholar] [CrossRef]
- Winder-Rhodes, S.E.; Garcia-Reitböck, P.; Ban, M.; Evans, J.R.; Jacques, T.S.; Kemppinen, A.; Foltynie, T.; Williams-Gray, C.H.; Chinnery, P.F.; Hudson, G.; et al. Genetic and pathological links between Parkinson’s disease and the lysosomal disorder Sanfilippo syndrome. Mov. Disord. 2012, 27, 312–315. [Google Scholar] [CrossRef]
- Cali, I.; Cohen, M.L.; Haik, S.; Parchi, P.; Giaccone, G.; Collins, S.J.; Kofskey, D.; Wang, H.; McLean, C.A.; Brandel, J.P.; et al. Iatrogenic Creutzfeldt-Jakob disease with Amyloid-β pathology: An international study. Acta Neuropathol. Commun. 2018, 6, 5. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W.; Quaranta, V.; Eanes, E.D. The amyloid deposits in Alzheimer’s disease: Their nature and pathogenesis. Appl. Pathol. 1984, 2, 357–369. [Google Scholar]
- Karmacharya, M.B.; Hada, B.; Park, S.R.; Choi, B.H. Low-Intensity Ultrasound Decreases α-Synuclein Aggregation via Attenuation of Mitochondrial Reactive Oxygen Species in MPP (+)-Treated PC12 Cells. Mol. Neurobiol. 2017, 54, 6235–6244. [Google Scholar] [CrossRef]
- Jiang, P.; Gan, M.; Yen, S.H.C. Dopamine prevents lipid peroxidation-induced accumulation of toxic α-synuclein oligomers by preserving autophagy-lysosomal function. Front. Cell. Neurosci. 2013, 7, 81. [Google Scholar] [CrossRef]
- Sinha, M.S.; Giraldo, A.M.V.; Öllinger, K.; Hallbeck, M.; Civitelli, L. Lipid vesicles affect the aggregation of 4-hydroxy-2-nonenal-modified α-synuclein oligomers. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 3060–3068. [Google Scholar] [CrossRef]
- Ischiropoulos, H. Oxidative modifications of alpha-synuclein. Ann. N. Y. Acad. Sci. 2003, 991, 93–100. [Google Scholar] [CrossRef]
- Yamin, G.; Uversky, V.N.; Fink, A.L. Nitration inhibits fibrillation of human α-synuclein in vitro by formation of soluble oligomers. FEBS Lett. 2003, 542, 147–152. [Google Scholar] [CrossRef]
- Souza, J.M. Dityrosine Cross-linking Promotes Formation of Stable alpha-Synuclein Polymers Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef]
- Hodara, R.; Norris, E.H.; Giasson, B.I.; Mishizen-Eberz, A.J.; Lynch, D.R.; Lee, V.M.; Ischiropoulos, H. Functional consequences of alpha-synuclein tyrosine nitration: Diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 2004, 279, 47746–47753. [Google Scholar] [CrossRef] [PubMed]
- Barrett, P.J.; Greenamyre, J.T. Post-translational modification of α-synuclein in Parkinson׳s disease. Brain Res. 2015, 1628, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.C.; König, S.; Roeber, S.; et al. Nitration of Tyrosine 10 Critically Enhances Amyloid β Aggregation and Plaque Formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Napoli, K.L.; Kahan, B.D. Nonselective measurement of cyclosporine for therapeutic drug monitoring by fluorescence polarization immunoassay with a rabbit polyclonal antibody: I. Evaluation of the serum methodology and comparison with a sheep polyclonal antibody in an 3H-tracer mediated radioimmunoassay. Transplant. Proc. 1990, 22, 1175–1180. [Google Scholar] [PubMed]
- Kostka, M.; Högen, T.; Danzer, K.M.; Levin, J.; Habeck, M.; Wirth, A.; Wagner, R.; Glabe, C.G.; Finger, S.; Heinzelmann, U.; et al. Single Particle Characterization of Iron-induced Pore-forming α-Synuclein Oligomers. J. Biol. Chem. 2008, 283, 10992–11003. [Google Scholar] [CrossRef]
- Van Bergen, J.M.G.; Li, X.; Hua, J.; Schreiner, S.J.; Steininger, S.C.; Quevenco, F.C.; Wyss, M.; Gietl, A.F.; Treyer, V.; Leh, S.E.; et al. Colocalization of cerebral iron with Amyloid beta in Mild Cognitive Impairment. Sci. Rep. 2016, 6, 35514. [Google Scholar] [CrossRef] [Green Version]
- Joppe, K.; Roser, A.E.; Maass, F.; Lingor, P. The Contribution of Iron to Protein Aggregation Disorders in the Central Nervous System. Front. Neurosci. 2019, 13, 15. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, L.; Zhang, L.; Peng, Y.; Zhou, F. Redox reactions of the α-synuclein-Cu2+ complex and their effects on neuronal cell viability. Biochemistry 2010, 49, 8134–8142. [Google Scholar] [CrossRef]
- Yang, L.; Wang, W.; Chen, J.; Wang, N.; Zheng, G. A comparative study of resveratrol and resveratrol-functional selenium nanoparticles: Inhibiting amyloid β aggregation and reactive oxygen species formation properties. J. Biomed. Mater. Res. Part A 2018, 106, 3034–3041. [Google Scholar] [CrossRef]
- Kikuchi, H.; Kuribayashi, F.; Kiwaki, N.; Takami, Y.; Nakayama, T. GCN5 Regulates the Superoxide-Generating System in Leukocytes Via Controlling gp91-phox Gene Expression. J. Immunol. 2011, 186, 3015–3022. [Google Scholar] [CrossRef] [Green Version]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Yang, G.; Zhang, X.; Wang, P.; Weng, X.; Yang, Y.; Li, Z.; Fang, M.; Xu, Y.; Sun, A.; et al. Megakaryocytic Leukemia 1 Bridges Epigenetic Activation of NADPH Oxidase in Macrophages to Cardiac Ischemia-Reperfusion Injury. Circulation 2018, 138, 2820–2836. [Google Scholar] [CrossRef]
- Patnala, R.; Arumugam, T.V.; Gupta, N.; Dheen, S.T. HDAC Inhibitor Sodium Butyrate-Mediated Epigenetic Regulation Enhances Neuroprotective Function of Microglia During Ischemic Stroke. Mol. Neurobiol. 2016, 54, 6391–6411. [Google Scholar] [CrossRef]
- Ni, J.; Wang, X.; Chen, S.; Liu, H.; Wang, Y.; Xu, X.; Cheng, J.; Jia, J.; Zhen, X. MicroRNA let-7c-5p protects against cerebral ischemia injury via mechanisms involving the inhibition of microglia activation. Brain Behav. Immun. 2015, 49, 75–85. [Google Scholar] [CrossRef]
- Lv, J.; Zeng, Y.; Qian, Y.; Dong, J.; Zhang, Z.; Zhang, J. MicroRNA let-7c-5p improves neurological outcomes in a murine model of traumatic brain injury by suppressing neuroinflammation and regulating microglial activation. Brain Res. 2018, 1685, 91–104. [Google Scholar] [CrossRef]
- Burguillos, M.A.; Deierborg, T.; Kavanagh, E.; Persson, A.; Hajji, N.; Garcia-Quintanilla, A.; Cano, J.; Brundin, P.; Englund, E.; Venero, J.L.; et al. Caspase signalling controls microglia activation and neurotoxicity. Nature 2011, 472, 319–324. [Google Scholar] [CrossRef]
- Yang, Z.; Zhong, L.; Zhong, S.; Xian, R.; Yuan, B. miR-203 protects microglia mediated brain injury by regulating inflammatory responses via feedback to MyD88 in ischemia. Mol. Immunol. 2015, 65, 293–301. [Google Scholar] [CrossRef]
- Yip, P.K.; Bowes, A.L.; E Hall, J.C.; A Burguillos, M.; Ip, T.H.R.; Baskerville, T.; Liu, Z.H.; Mohamed, M.A.E.K.; Getachew, F.; Lindsay, A.D.; et al. Docosahexaenoic acid reduces microglia phagocytic activity via miR-124 and induces neuroprotection in rodent models of spinal cord contusion injury. Hum. Mol. Genet. 2019, 28, 2427–2448. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Mandrekar-Colucci, S.; Hall, J.C.; Sweet, D.R.; Schmitt, P.J.; Xu, X.; Guan, Z.; Mo, X.; Guerau-De-Arellano, M.; Popovich, P.G. miR-155 Deletion in Mice Overcomes Neuron-Intrinsic and Neuron-Extrinsic Barriers to Spinal Cord Repair. J. Neurosci. 2016, 36, 8516–8532. [Google Scholar] [CrossRef] [Green Version]
- Gaudet, A.D.; Fonken, L.K.; Watkins, L.R.; Nelson, R.J.; Popovich, P.G. MicroRNAs: Roles in Regulating Neuroinflammation. Neuroscientist 2017, 24, 221–245. [Google Scholar] [CrossRef] [Green Version]
- Von Bernhardi, R.; Bernhardi, L.E.V.; Eugenin, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [Green Version]
- Sierra, A.; McEwen, B.S.; Bulloch, K.; Gottfried-Blackmore, A.C.; Gottfried-Blackmore, A.C. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef]
- Ashapkin, V.V.; Kutueva, L.I.; Vanyushin, B.F. Aging as an Epigenetic Phenomenon. Curr. Genom. 2017, 18, 385–407. [Google Scholar] [CrossRef] [Green Version]
- Julien, C.; Tremblay, C.; Émond, V.; Lebbadi, M.; Salem, N.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef]
- Lutz, M.I.; Milenkovic, I.; Regelsberger, G.; Kovacs, G.G. Distinct Patterns of Sirtuin Expression During Progression of Alzheimer’s Disease. Neuromol. Med. 2014, 16, 405–414. [Google Scholar] [CrossRef]
- Cho, S.H.; Chen, J.A.; Sayed, F.; Ward, M.E.; Gao, F.; Nguyen, T.A.; Krabbe, G.; Sohn, P.D.; Lo, I.; Minami, S.; et al. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. J. Neurosci. 2015, 35, 807–818. [Google Scholar] [CrossRef]
- Tang, Y.; Li, T.; Li, J.; Yang, J.; Liu, H.; Zhang, X.J.; Le, W. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 2013, 21, 369–380. [Google Scholar] [CrossRef]
- Jucaite, A.; Svenningsson, P.; Rinne, J.O.; Cselényi, Z.; Varnäs, K.; Johnström, P.; Amini, N.; Kirjavainen, A.; Helin, S.; Minkwitz, M.; et al. Effect of the myeloperoxidase inhibitor AZD3241 on microglia: A PET study in Parkinson’s disease. Brain 2015, 138, 2687–2700. [Google Scholar] [CrossRef]
- Rees, K.; Stowe, R.; Patel, S.; Ives, N.; Breen, K.; Clarke, C.E.; Ben-Shlomo, Y.; Ben-Shlomo, Y. Non-steroidal anti-inflammatory drugs as disease-modifying agents for Parkinson’s disease: Evidence from observational studies. Cochrane Database Syst. Rev. 2011. [Google Scholar] [CrossRef]
- Clarke, C.E.; Speller, J.M.; Clarke, J.A. Pramipexole versus bromocriptine for levodopa-induced complications in Parkinson’s disease. Cochrane Database Syst. Rev. 2000. [Google Scholar] [CrossRef]
- Clarke, C.E.; Speller, J.M. Pergolide for levodopa-induced complications in Parkinson’s disease. Cochrane Database Syst. Rev. 2000. [Google Scholar] [CrossRef]
- Ramaker, C.; Hilten, J. Bromocriptine/levodopa combined versus levodopa alone for early Parkinson’s disease. Cochrane Database Syst. Rev. 2002. [Google Scholar] [CrossRef]
- Clarke, C.E.; Deane, K.H. Cabergoline versus bromocriptine for levodopa-induced complications in Parkinson’s disease. Cochrane Database Syst. Rev. 2001. [Google Scholar] [CrossRef]
- Clarke, C.E.; Deane, K.H. Ropinirole versus bromocriptine for levodopa-induced complications in Parkinson’s disease. Cochrane Database Syst. Rev. 2000. [Google Scholar] [CrossRef]
- Caslake, R.; MacLeod, A.; Ives, N.; Stowe, R.; Counsell, C. Monoamine oxidase B inhibitors versus other dopaminergic agents in early Parkinson’s disease. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]
- Investigators, N.N.P. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology 2006, 66, 664–671. [Google Scholar] [CrossRef]
- Farina, N.; Llewellyn, D.; Isaac, M.G.E.K.N.; Tabet, N. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Syst. Rev. 2017. [Google Scholar] [CrossRef]
- López-Arrieta, J.M.; Rodríguez, J.L.; Sanz, F.J.F.; López-Arrieta, J.; López-Arrieta, J. Nicotine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2001. [Google Scholar] [CrossRef]
- Flicker, L.; Grimley Evans, G. Piracetam for dementia or cognitive impairment. Cochrane Database Syst. Rev. 2001. [Google Scholar] [CrossRef]
- Mccleery, J.; Abraham, R.P.; Denton, D.A.; Rutjes, A.W.; Chong, L.Y.; Al-Assaf, A.S.; Griffith, D.J.; Rafeeq, S.; Yaman, H.; Malik, M.A.; et al. Vitamin and mineral supplementation for preventing dementia or delaying cognitive decline in people with mild cognitive impairment. Cochrane Database Syst. Rev. 2018. [Google Scholar] [CrossRef]
- Tabet, N.; Feldman, H. Ibuprofen for Alzheimer’s disease. Cochrane Database Syst. Rev. 2003. [Google Scholar] [CrossRef]
- Tabet, N.; Feldman, H. Indomethacin for the treatment of Alzheimer’s disease patients. Cochrane Database Syst. Rev. 2002. [Google Scholar] [CrossRef]
- Jaturapatporn, D.; Isaac, M.G.E.K.N.; Mccleery, J.; Tabet, N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Metz, L.M.; Li, D.K.; Traboulsee, A.L.; Duquette, P.; Eliasziw, M.; Cerchiaro, G.; Greenfield, J.; Riddehough, A.; Yeung, M.; Kremenchutzky, M.; et al. Trial of Minocycline in a Clinically Isolated Syndrome of Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 2122–2133. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Luo, M.; Zhang, X.; Luo, H. Statins for multiple sclerosis. Cochrane Database Syst. Rev. 2010. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, F.; Sun, F.; Gu, K.; Dong, S.; He, D. Dimethyl fumarate for multiple sclerosis. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef]
- He, D.; Han, K.; Gao, X.; Dong, S.; Feng, Z.; Wu, S. Laquinimod for multiple sclerosis. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Pucci, E.; Branãs, P.; D’Amico, R.; Giuliani, G.; Solari, A.; Taus, C.; Tato, P.B. Amantadine for fatigue in multiple sclerosis. Cochrane Database Syst. Rev. 2007. [Google Scholar] [CrossRef]
- La Mantia, L.; Tramacere, I.; Firwana, B.; Pacchetti, I.; Palumbo, R.; Filippini, G. Fingolimod for relapsing-remitting multiple sclerosis. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef]
- Jagannath, V.A.; Filippini, G.; Di Pietrantonj, C.; Asokan, G.V.; Robak, E.W.; Whamond, L.; Robinson, S.A. Vitamin D for the management of multiple sclerosis. Cochrane Database Syst. Rev. 2018. [Google Scholar] [CrossRef]
- Tejani, A.M.; Wasdell, M.; Spiwak, R.; Rowell, G.; Nathwani, S. Carnitine for fatigue in multiple sclerosis. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Farinotti, M.; Vacchi, L.; Simi, S.; Di Pietrantonj, C.; Brait, L.; Filippini, G. Dietary interventions for multiple sclerosis. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Pastula, D.M.; Moore, D.H.; Bedlack, R.S. Creatine for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst. Rev. 2010. [Google Scholar] [CrossRef]
- Miller, R.G.; Mitchell, J.D.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Ahmadi, M.; Agah, E.; Nafissi, S.; Jaafari, M.R.; Harirchian, M.H.; Sarraf, P.; Faghihi-Kashani, S.; Hosseini, S.J.; Ghoreishi, A.; Aghamollaii, V.; et al. Safety and Efficacy of Nanocurcumin as Add-On Therapy to Riluzole in Patients with Amyotrophic Lateral Sclerosis: A Pilot Randomized Clinical Trial. Neurotherapeutics 2018, 15, 430–438. [Google Scholar] [CrossRef]
- Orrell, R.W.; Lane, R.J.; Ross, M. Antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst. Rev. 2005. [Google Scholar] [CrossRef]
- Bender, A.; Klopstock, T. Creatine for neuroprotection in neurodegenerative disease: end of story? Amino Acids 2016, 48, 1929–1940. [Google Scholar] [CrossRef]
- Dobson, L.; Träger, U.; Farmer, R.; Hayardeny, L.; Loupe, P.; Hayden, M.R.; Tabrizi, S.J. Laquinimod dampens hyperactive cytokine production in Huntington’s disease patient myeloid cells. J. Neurochem. 2016, 137, 782–794. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, S.M.; Hernán, M.A.; Schwarzschild, M.A.; Willett, W.C.; Colditz, G.A.; Speizer, F.E.; Ascherio, A. Nonsteroidal Anti-inflammatory Drugs and the Risk of Parkinson Disease. Arch. Neurol. 2003, 60, 1059–1064. [Google Scholar] [CrossRef] [Green Version]
- Anthony, J.C.; Breitner, J.C.S.; Zandi, P.P.; Meyer, M.R.; Jurasova, I.; Norton, M.C.; Stone, S.V. Reduced prevalence of AD in users of NSAIDs and H2 receptor antagonists: The Cache County Study. Neurology 2000, 54, 2066–2071. [Google Scholar] [CrossRef]
- Yong, V.W.; Wells, J.; Giuliani, F.; Casha, S.; Power, C.; Metz, L.M. The promise of minocycline in neurology. Lancet Neurol. 2004, 3, 744–751. [Google Scholar] [CrossRef]
- Garrido-Mesa, N.; Zarzuelo, A.; Galvez, J. Minocycline: Far beyond an antibiotic. Br. J. Pharmacol. 2013, 169, 337–352. [Google Scholar] [CrossRef]
- Sapadin, A.N.; Fleischmajer, R. Tetracyclines: Nonantibiotic properties and their clinical implications. J. Am. Acad. Dermatol. 2006, 54, 258–265. [Google Scholar] [CrossRef]
- Soory, M. A Role for Non-Antimicrobial Actions of Tetracyclines in Combating Oxidative Stress in Periodontal and Metabolic Diseases: A Literature Review. Open Dent. J. 2008, 2, 5–12. [Google Scholar] [CrossRef]
- Griffin, M.O.; Fricovsky, E.; Ceballos, G.; Villarreal, F. Tetracyclines: A pleitropic family of compounds with promising therapeutic properties. Review of the literature. Am. J. Physiol. Cell Physiol. 2010, 299, 539–548. [Google Scholar] [CrossRef]
- Blum, D.; Chtarto, A.; Tenenbaum, L.; Brotchi, J.; Levivier, M. Clinical potential of minocycline for neurodegenerative disorders. Neurobiol. Dis. 2004, 17, 359–366. [Google Scholar] [CrossRef]
- Li, C.; Yuan, K.; Schluesener, H. Impact of minocycline on neurodegenerative diseases in rodents: A meta-analysis. Rev. Neurosci. 2013, 24, 553–562. [Google Scholar] [CrossRef]
- Du, Y.; Ma, Z.; Lin, S.; Dodel, R.C.; Gao, F.; Bales, K.R.; Triarhou, L.C.; Chernet, E.; Perry, K.W.; Nelson, D.L.G.; et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 14669–14674. [Google Scholar] [CrossRef]
- Cankaya, S.; Cankaya, B.; Kilic, U.; Kilic, E.; Yulug, B. The therapeutic role of minocycline in Parkinson’s disease. Drugs Context 2019, 8, 212553. [Google Scholar] [CrossRef]
- Jacob, K.D.; Hooten, N.N.; Trzeciak, A.R.; Evans, M.K. Markers of oxidant stress that are clinically relevant in aging and age-related disease. Mech. Ageing Dev. 2013, 134, 139–157. [Google Scholar] [CrossRef] [Green Version]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxidative Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef]
- Winterbourn, C.C. The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1840, 730–738. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef]
- Gomes, A.; Fernandes, E.; Lima, J.L. Fluorescence probes used for detection of reactive oxygen species. J. Biochem. Biophys. Methods 2005, 65, 45–80. [Google Scholar] [CrossRef]
- Khan, J.; Brennan, M.D.; Bradley, N.; Gao, B.; Bruckdorfer, R.; Jacobs, M. 3-Nitrotyrosine in the proteins of human plasma determined by an ELISA method. Biochem. J. 1998, 330, 795–801. [Google Scholar] [CrossRef]
- Teixeira, D.; Fernandes, R.; Prudêncio, C.; Vieira, M. 3-Nitrotyrosine quantification methods: Current concepts and future challenges. Biochimie 2016, 125, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ito, F.; Sono, Y.; Ito, T. Measurement and Clinical Significance of Lipid Peroxidation as a Biomarker of Oxidative Stress: Oxidative Stress in Diabetes, Atherosclerosis, and Chronic Inflammation. Antioxidants 2019, 8, 72. [Google Scholar] [CrossRef]
- Sekhar, R.V.; Patel, S.G.; Guthikonda, A.P.; Reid, M.; Balasubramanyam, A.; Taffet, G.E.; Jahoor, F. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Am. J. Clin. Nutr. 2011, 94, 847–853. [Google Scholar] [CrossRef] [Green Version]
- Ho, E.; Galougahi, K.K.; Liu, C.C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [Green Version]
- Tipple, T.E.; Rogers, L.K. Methods for the determination of plasma or tissue glutathione levels. Breast Cancer 2012, 889, 315–324. [Google Scholar]
- Weber, D.; Davies, M.J.; Grune, T. Determination of protein carbonyls in plasma, cell extracts, tissue homogenates, isolated proteins: Focus on sample preparation and derivatization conditions. Redox Biol. 2015, 5, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Tammen, H.; Schulte, I.; Hess, R.; Menzel, C.; Kellmann, M.; Mohring, T.; Schulz-Knappe, P.; Schulz-Knappe, P. Peptidomic analysis of human blood specimens: Comparison between plasma specimens and serum by differential peptide display. Proteomics 2005, 5, 3414–3422. [Google Scholar] [CrossRef]
- Delgado-Andrade, C. Carboxymethyl-lysine: Thirty years of investigation in the field of AGE formation. Food Funct. 2016, 7, 46–57. [Google Scholar] [CrossRef]
- Colzani, M.; Aldini, G.; Carini, M. Mass spectrometric approaches for the identification and quantification of reactive carbonyl species protein adducts. J. Proteom. 2013, 92, 28–50. [Google Scholar] [CrossRef]
- Schleicher, E.; Wieland, O.H. Specific Quantitation by HPLC of Protein (Lysine) Bound Glucose in Human Serum Albumin and Other Glycosylated Proteins. Clin. Chem. Lab. Med. 1981, 19, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Gaut, J.P.; Byun, J.; Tran, H.D.; Heinecke, J.W. Artifact-Free Quantification of Free 3-Chlorotyrosine, 3-Bromotyrosine, and 3-Nitrotyrosine in Human Plasma by Electron Capture–Negative Chemical Ionization Gas Chromatography Mass Spectrometry and Liquid Chromatography–Electrospray Ionization Tandem Mass Spectrometry. Anal. Biochem. 2002, 300, 252–259. [Google Scholar]
- Taylor, E.L.; Armstrong, K.R.; Perrett, D.; Hattersley, A.T.; Winyard, P.G. Optimisation of an Advanced Oxidation Protein Products Assay: Its Application to Studies of Oxidative Stress in Diabetes Mellitus. Oxidative Med. Cell. Longev. 2015, 2015, 496271. [Google Scholar] [CrossRef]
- Sousa, B.C.; Pitt, A.R.; Spickett, C.M. Chemistry and analysis of HNE and other prominent carbonyl-containing lipid oxidation compounds. Free Radic. Biol. Med. 2017, 111, 294–308. [Google Scholar] [CrossRef]
- Domijan, A.M.; Ralić, J.; Brkanac, S.R.; Rumora, L.; Žanić-Grubišić, T.; Žanić-Grubišić, T. Quantification of malondialdehyde by HPLC-FL—Application to various biological samples. Biomed. Chromatogr. 2014, 29, 41–46. [Google Scholar] [CrossRef]
- Milne, G.L.; Dai, Q.; Roberts, L.J. The isoprostanes—25 years later. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2014, 1851, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Milne, G.L.; Sanchez, S.C.; Musiek, E.S.; Morrow, J.D. Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nat. Protoc. 2007, 2, 221–226. [Google Scholar] [CrossRef]
- Li, W.; Laird, J.M.; Lu, L.; Roychowdhury, S.; Nagy, L.E.; Zhou, R.; Crabb, J.W.; Salomon, R.G. Isolevuglandins covalently modify phosphatidylethanolamines in vivo: Detection and quantitative analysis of hydroxylactam adducts. Free Radic. Biol. Med. 2009, 47, 1539–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dizdaroglu, M.; Jaruga, P.; Birincioglu, M.; Rodriguez, H. Free radical-induced damage to DNA: Mechanisms and measurement. Free Radic. Biol. Med. 2002, 32, 1102–1115. [Google Scholar] [CrossRef]
- Weimann, A.; Belling, D.; Poulsen, H.E. Quantification of 8-oxo-guanine and guanine as the nucleobase, nucleoside and deoxynucleoside forms in human urine by high-performance liquid chromatography–electrospray tandem mass spectrometry. Nucleic Acids Res. 2002, 30, e7. [Google Scholar] [CrossRef]
- Cooke, M.; Olinski, R.; Loft, S. Measurement and Meaning of Oxidatively Modified DNA Lesions in Urine. Cancer Epidemiol. Biomark. Prev. 2008, 17, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Weimann, A.; Broedbaek, K.; Henriksen, T.; Stovgaard, E.S.; Poulsen, H.E. Assays for urinary biomarkers of oxidatively damaged nucleic acids. Free Radic. Res. 2012, 46, 531–540. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Compound | Effects | Reference |
|---|---|---|---|
| Huntington´s disease | Minocycline | Trial in progress | CN-01508775 |
| Laquinimod | Trial in progress | CN-01364776 | |
| AZD3241 | Trial in progress | [188] | |
| Riluzole | Trial in progress | CN-01513126 | |
| Antioxidants | Trials in progress | CN-00120692;CN-00150820 | |
| Fenifibrate | Trial in progress | CN-01574117 | |
| Memantine | Trial in progress | CN-01533522 | |
| Creatin | Trial in progress | NCT00592995 | |
| Parkinson’s disease | Green tea | Trial in progress | NCT00461942 |
| Polyphenols | Trial in progress | NCT00461942 | |
| NSAIDs | May reduce the risk of developing PD | [189] | |
| Q10 coenzyme | Trial in progress | NCT00076492 | |
| Pramipexole | Improved motor impairments and disability | [190] | |
| Pergolide | Improved motor impairments and disability | [191] | |
| Bromocriptine | No prevention in the onset of motor complications | [192] | |
| Cabergoline | Improved motor impairments and disability | [193] | |
| Ropinirole | Improved motor impairments and disability | [194] | |
| MAO B inhibitors | Reduced rate of motor fluctuations | [195] | |
| Minocycline | Phase II trial | [196] | |
| Polyphenols | Trials in progress | NCT01001637; NCT00743743; NCT00164749 | |
| Vitamin E | No evidence | [197] | |
| Nicotine | No results available | [198] | |
| Piracetam | No benefit was shown | [199] | |
| Vitamins and minerals | Evidence very limited | [200] | |
| Masitinib | Trial in progress | CN-01867921 | |
| NSAIDs | Sparing effect ranging between 36% and 80% | [201,202,203] | |
| Minocycline | Trial in progress | CN-01797324; CN-01847636 | |
| Multiple sclerosis | Minocycline | Decreased risk of conversion from a clinically isolate syndrome to multiple sclerosis | [204] |
| Statins | No convincing evidence | [205] | |
| Dimethyl Fumarate | Moderate-quality evidence of decreased relapses frequency | [206] | |
| Laquinimod | Low-level evidence as a disease-modifying therapy | [207] | |
| Amantadine | Poorly documented | [208] | |
| Fingolimod | Reduced inflammatory disease activity | [209] | |
| Vitamin D | No benefit | [210] | |
| Carnitine | Insufficient evidence | [211] | |
| PUFAs | Reduced relapses frequency | [212]; CN-01579873 | |
| Lipoic acid | Trials in progress | CN-01518782; CN-01878397; CN-01266929 | |
| Epigallocatechin gallate | Trial in progress | CN-01794896 | |
| Q10 coenzyme | Trials in progress | CN-00866966; CN-01136521 | |
| Melatonin | Trial in progress | CN-01114089 | |
| Crocin | Trial in progress | CN-01836077 | |
| Curcumin | Trial in progress | N-01896018 | |
| Alpha-tocopherol | Trial in progress | CN-00912436 | |
| ω3 fatty acids | Trial in progress | CN-01650553 | |
| Fish oil | Trial in progress | CN-00982838 | |
| Creatin | Trial in progress | CN-01502285 | |
| NSAIDs | Trial in progress | CN-01010452 | |
| Amyotrophic lateral sclerosis | Vitamin E | Trial in progress | CN-00442211 |
| Creatin | No effect on survival | [213] | |
| Minocyclin | Trials in progress | CN-01616430; CN-01259752; CN-01381473 | |
| CN-00730827 | |||
| Riluzole | Probably prolongs survival | [214] | |
| Celecoxib | Trial in progress | CN-00566124 | |
| Curcumin | Might improve survival probability | [215] | |
| Antioxidants | Insufficient evidence of efficacy | [216] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Revilla, J.; Alonso-Bellido, I.M.; Burguillos, M.A.; Herrera, A.J.; Espinosa-Oliva, A.M.; Ruiz, R.; Cruz-Hernández, L.; García-Domínguez, I.; Roca-Ceballos, M.A.; Santiago, M.; et al. Reformulating Pro-Oxidant Microglia in Neurodegeneration. J. Clin. Med. 2019, 8, 1719. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8101719
García-Revilla J, Alonso-Bellido IM, Burguillos MA, Herrera AJ, Espinosa-Oliva AM, Ruiz R, Cruz-Hernández L, García-Domínguez I, Roca-Ceballos MA, Santiago M, et al. Reformulating Pro-Oxidant Microglia in Neurodegeneration. Journal of Clinical Medicine. 2019; 8(10):1719. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8101719
Chicago/Turabian StyleGarcía-Revilla, Juan, Isabel M. Alonso-Bellido, Miguel A. Burguillos, Antonio J. Herrera, Ana M. Espinosa-Oliva, Rocío Ruiz, Luis Cruz-Hernández, Irene García-Domínguez, María A. Roca-Ceballos, Marti Santiago, and et al. 2019. "Reformulating Pro-Oxidant Microglia in Neurodegeneration" Journal of Clinical Medicine 8, no. 10: 1719. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8101719