Liposomal Lapatinib in Combination with Low-Dose Photodynamic Therapy for the Treatment of Glioma

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Liposomal Encapsulation of Lapatinib
2.2. Quantification of Encapsulated Lapatinib
2.3. Physical Characterization of Liposomal Lapatinib
2.4. Cell Lines
2.5. Live-Cell Imaging of Glioma Cell Lines
2.6. In Vitro PDT
2.7. Inoculation of Tumours
2.8. MRI Methods
2.9. In Vivo PDT
2.10. Histology
3. Results
3.1. Liposomal Encapsulation of Lapatinib
3.2. Live-Cell Imaging
3.3. In Vitro PDT
3.4. In Vivo PDT
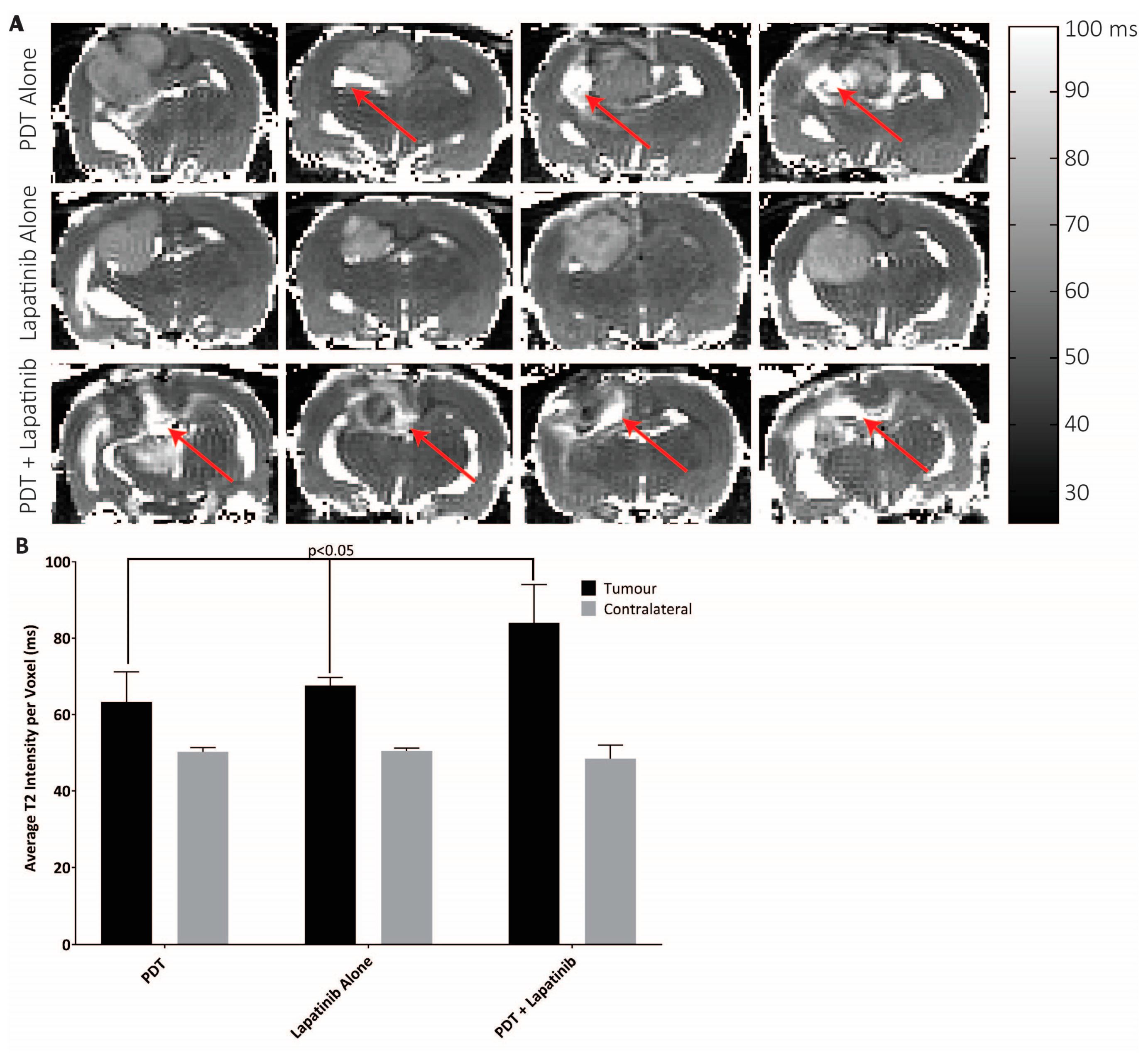
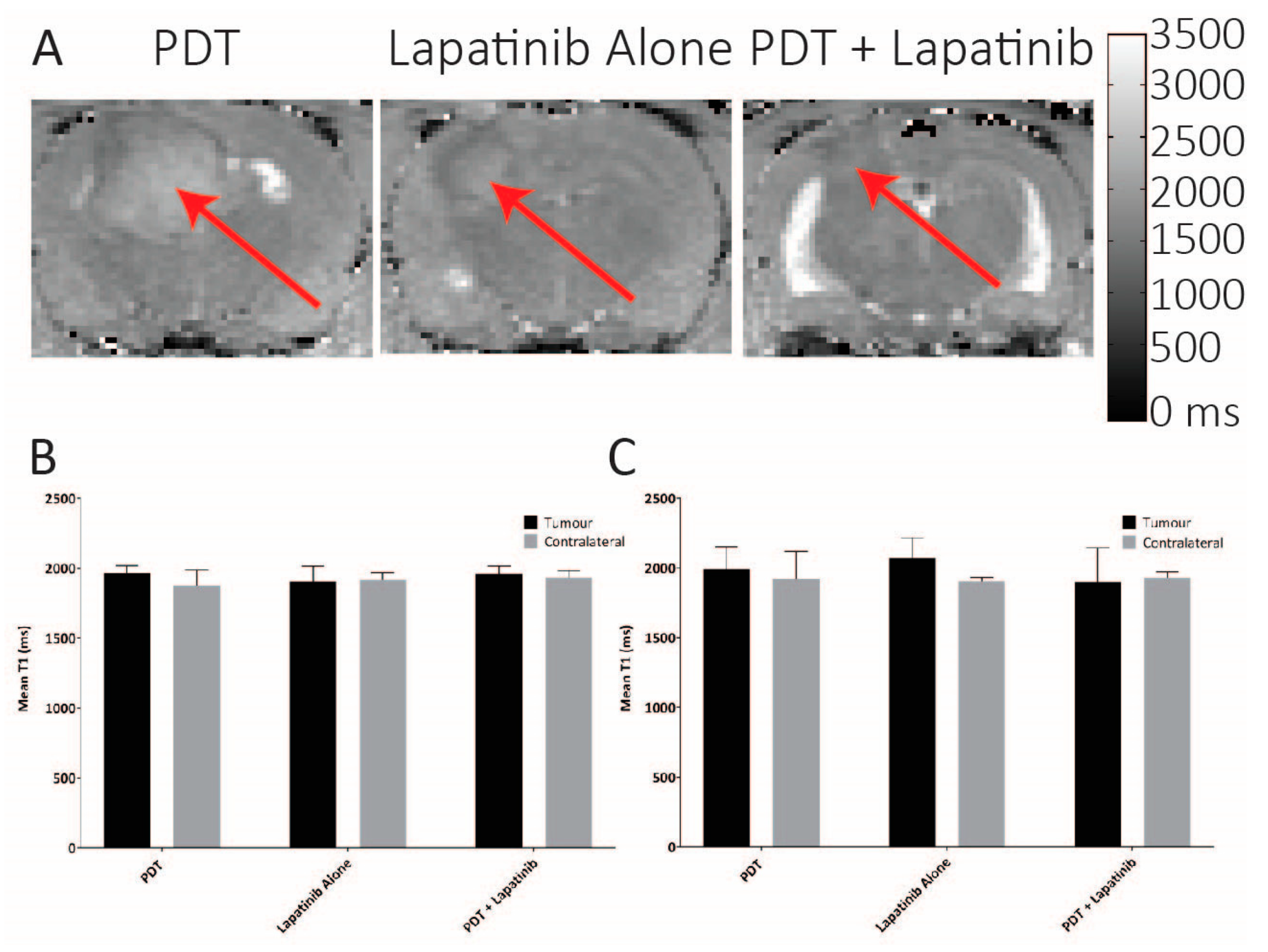
3.5. Quantitative MRI
4. Discussion
4.1. Key Points:
- Lapatinib in conjunction with PDT improves efficacy in glioma;
- Lapatinib increases PpIX concentrations in vivo to increased contrast for fluorescence-guided resection (FGR).
4.2. Importance of the Study:
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCG2 | ATP Binding Cassette Subfamily G Member 2; |
| ALA | aminolevulinic acid; |
| CBF | cerebral blood flow; |
| CTV | Clinical Target Volume; |
| DOTAP | ; 1,2-dioleoyl-3-trimethylammonium-propane; |
| DPPC | 1,2-dipalmitoyl-snglycero-3-phosphocholine; |
| DSPE-mPEG | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol) |
| EGFR | Epidermal Growth Factor Receptor; |
| FAIR | flow alternating inversion recovery; |
| FDA | Food and Drug Administration; |
| FGR | fluorescence Guided-Resection; |
| GBM | Glioblastoma Multiforme; |
| HBEGF | Heparin-binding EGF-like growth factor; |
| HGG | High-Grade Glioma; |
| LED | Light emitting diode; |
| MRI | Magnetic Resonance Imaging; |
| MTR | MitoTracker Red;. |
| PDT | Photodynamic Therapy; |
| PpIX | Protoporphyrin IX; |
| RARE | rapid acquisition with relaxation enhancement; |
| ROI | Region of Interest |
| SCID | Severe Combined Immunodeficient; |
| TKI | Tyrosine Kinase Inhibitor; |
References
- Beije, N.; Kraan, J.; Taal, W.; van der Holt, B.; Oosterkamp, H.M.; Walenkamp, A.M.; Beerepoot, L.; Hanse, M.; van Linde, M.E.; Otten, A.; et al. Prognostic value and kinetics of circulating endothelial cells in patients with recurrent glioblastoma randomised to bevacizumab plus lomustine, bevacizumab single agent or lomustine single agent. A report from the Dutch Neuro-Oncology Group BELOB trial. Br. J. Cancer 2015, 113, 226–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutledge, M.R.; Waddell, J.A.; Solimando, D.A., Jr. Bevacizumab and Temozolomide Plus Radiation Regimen for Glioblastoma Multiforme. Hosp. Pharm. 2015, 50, 672–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saran, F.; Chinot, O.L.; Henriksson, R.; Mason, W.; Wick, W.; Cloughesy, T.; Dhar, S.; Pozzi, E.; Garcia, J.; Nishikawa, R. Bevacizumab, temozolomide, and radiotherapy for newly diagnosed glioblastoma: Comprehensive safety results during and after first-line therapy. Neuro Oncol. 2016, 18, 991–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abla, A.A.; Rutledge, W.C.; Seymour, Z.A.; Guo, D.; Kim, H.; Gupta, N.; Sneed, P.K.; Barani, I.J.; Larson, D.; McDermott, M.W.; et al. A treatment paradigm for high-grade brain arteriovenous malformations: Volume-staged radiosurgical downgrading followed by microsurgical resection. J. Neurosurg. 2015, 122, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Crowley, R.W.; Pouratian, N.; Sheehan, J.P. Gamma knife surgery for glioblastoma multiforme. Neurosurg. Focus 2006, 20, E17. [Google Scholar] [CrossRef]
- Larson, E.W.; Peterson, H.E.; Lamoreaux, W.T.; MacKay, A.R.; Fairbanks, R.K.; Call, J.A.; Carlson, J.D.; Ling, B.C.; Demakas, J.J.; Cooke, B.S.; et al. Clinical outcomes following salvage Gamma Knife radiosurgery for recurrent glioblastoma. World J. Clin. Oncol. 2014, 5, 142–148. [Google Scholar] [CrossRef]
- Alan Mitteer, R.; Wang, Y.; Shah, J.; Gordon, S.; Fager, M.; Butter, P.P.; Jun Kim, H.; Guardiola-Salmeron, C.; Carabe-Fernandez, A.; Fan, Y. Proton beam radiation induces DNA damage and cell apoptosis in glioma stem cells through reactive oxygen species. Sci. Rep. 2015, 5, 13961. [Google Scholar] [CrossRef] [Green Version]
- Amelio, D.; Lorentini, S.; Schwarz, M.; Amichetti, M. Intensity-modulated radiation therapy in newly diagnosed glioblastoma: A systematic review on clinical and technical issues. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2010, 97, 361–369. [Google Scholar] [CrossRef]
- Mizumoto, M.; Yamamoto, T.; Takano, S.; Ishikawa, E.; Matsumura, A.; Ishikawa, H.; Okumura, T.; Sakurai, H.; Miyatake, S.; Tsuboi, K. Long-term survival after treatment of glioblastoma multiforme with hyperfractionated concomitant boost proton beam therapy. Pract. Radiat. Oncol. 2015, 5, e9–e16. [Google Scholar] [CrossRef]
- Karavasilis, V.; Kotoula, V.; Pentheroudakis, G.; Televantou, D.; Lambaki, S.; Chrisafi, S.; Bobos, M.; Fountzilas, G. A phase I study of temozolomide and lapatinib combination in patients with recurrent high-grade gliomas. J. Neurol. 2013, 260, 1469–1480. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J.; et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stummer, W.; Stepp, H.; Moller, G.; Ehrhardt, A.; Leonhard, M.; Reulen, H.J. Technical principles for protoporphyrin-IX-fluorescence guided microsurgical resection of malignant glioma tissue. Acta Neurochir. 1998, 140, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Kostron, H.; Fiegele, T.; Akatuna, E. Combination of FOSCAN® mediated fluorescence guided resection and photodynamic treatment as new therapeutic concept for malignant brain tumors. Med Laser Appl. 2006, 21, 285–290. [Google Scholar] [CrossRef]
- Eljamel, S.; Petersen, M.; Valentine, R.; Buist, R.; Goodman, C.; Moseley, H. Comparison of intraoperative fluorescence and MRI image guided neuronavigation in malignant brain tumours, a prospective controlled study. Photodiagnosis Photodyn. Ther. 2013, 10, 356–361. [Google Scholar] [CrossRef]
- Eljamel, S. Photodynamic applications in brain tumors: A comprehensive review of the literature. Photodiagnosis Photodyn. Ther. 2010, 7, 76–85. [Google Scholar] [CrossRef]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Johansson, A.; Faber, F.; Kniebuhler, G.; Stepp, H.; Sroka, R.; Egensperger, R.; Beyer, W.; Kreth, F.W. Protoporphyrin IX fluorescence and photobleaching during interstitial photodynamic therapy of malignant gliomas for early treatment prognosis. Lasers Surg. Med. 2013, 45, 225–234. [Google Scholar] [CrossRef]
- Muragaki, Y.; Akimoto, J.; Maruyama, T.; Iseki, H.; Ikuta, S.; Nitta, M.; Maebayashi, K.; Saito, T.; Okada, Y.; Kaneko, S.; et al. Phase II clinical study on intraoperative photodynamic therapy with talaporfin sodium and semiconductor laser in patients with malignant brain tumors. J. Neurosurg. 2013, 119, 845–852. [Google Scholar] [CrossRef]
- Postiglione, I.; Chiaviello, A.; Aloj, S.M.; Palumbo, G. 5-aminolaevulinic acid/photo-dynamic therapy and gefitinib in non-small cell lung cancer cell lines: A potential strategy to improve gefitinib therapeutic efficacy. Cell Prolif. 2013, 46, 382–395. [Google Scholar] [CrossRef]
- Gallagher-Colombo, S.M.; Miller, J.; Cengel, K.A.; Putt, M.E.; Vinogradov, S.A.; Busch, T.M. Erlotinib Pretreatment Improves Photodynamic Therapy of Non–Small Cell Lung Carcinoma Xenografts via Multiple Mechanisms. Cancer Res. 2015, 75, 3118–3126. [Google Scholar] [CrossRef] [Green Version]
- Edmonds, C.; Hagan, S.; Gallagher-Colombo, S.M.; Busch, T.M.; Cengel, K.A. Photodynamic therapy activated signaling from epidermal growth factor receptor and STAT3: Targeting survival pathways to increase PDT efficacy in ovarian and lung cancer. Cancer Biol. Ther. 2012, 13, 1463–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilge, L.; Molpus, K.; Hasan, T.; Wilson, B.C. Light dosimetry for intraperitoneal photodynamic therapy in a murine xenograft model of human epithelial ovarian carcinoma. Photochem. Photobiol. 1998, 68, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Del Carmen, M.G.; Rizvi, I.; Chang, Y.; Moor, A.C.; Oliva, E.; Sherwood, M.; Pogue, B.; Hasan, T. Synergism of epidermal growth factor receptor-targeted immunotherapy with photodynamic treatment of ovarian cancer in vivo. J. Natl. Cancer Inst. 2005, 97, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Mallidi, S.; Liu, J.; Chiang, C.-T.; Mai, Z.; Goldschmidt, R.; Ebrahim-Zadeh, N.; Rizvi, I.; Hasan, T. Photodynamic therapy synergizes with irinotecan to overcome compensatory mechanisms and improve treatment outcomes in pancreatic cancer. Cancer Res. 2016, 76, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Kajimoto, Y.; Inoue, H.; Miyatake, S.; Ishikawa, T.; Kuroiwa, T. Gefitinib enhances the efficacy of photodynamic therapy using 5-aminolevulinic acid in malignant brain tumor cells. Photodiagnosis Photodyn. Ther. 2013, 10, 42–50. [Google Scholar] [CrossRef]
- Anand, S.; Ortel, B.J.; Pereira, S.P.; Hasan, T.; Maytin, E.V. Biomodulatory approaches to photodynamic therapy for solid tumors. Cancer Lett. 2012, 326, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Fisher, C.J.; Niu, C.J.; Lai, B.; Chen, Y.; Kuta, V.; Lilge, L.D. Modulation of PPIX synthesis and accumulation in various normal and glioma cell lines by modification of the cellular signaling and temperature. Lasers Surg. Med. 2013, 45, 460–468. [Google Scholar] [CrossRef]
- Thiessen, B.; Stewart, C.; Tsao, M.; Kamel-Reid, S.; Schaiquevich, P.; Mason, W.; Easaw, J.; Belanger, K.; Forsyth, P.; McIntosh, L.; et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2010, 65, 353–361. [Google Scholar] [CrossRef]
- Tangutoori, S.; Spring, B.Q.; Mai, Z.; Palanisami, A.; Mensah, L.; Hasan, T. Simultaneous delivery of cytotoxic and biologic therapeutics using nanophotoactivatable liposomes enhances treatment efficacy in a mouse model of pancreatic cancer. Nanomedicine 2016, 12, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.G.; Parker Kerrigan, B.C.; Hossain, A.; Gumin, J.; Shinojima, N.; Nwajei, F.; Ezhilarasan, R.; Love, P.; Sulman, E.P.; Lang, F.F. Ionizing radiation augments glioma tropism of mesenchymal stem cells. J. Neurosurg. 2018, 128, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Balvers, R.K.; Dirven, C.M.; Leenstra, S.; Lamfers, M.L. Malignant Glioma In Vitro Models: On the Utilization of Stem-like Cells. Curr. Cancer Drug Targets 2017, 17, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Stepp, H.; Beck, T.; Pongratz, T.; Meinel, T.; Kreth, F.W.; Tonn, J.; Stummer, W. ALA and malignant glioma: Fluorescence-guided resection and photodynamic treatment. J. Environ. Pathol. Toxicol. Oncol. Off. Organ Int. Soc. Environ. Toxicol. Cancer 2007, 26, 157–164. [Google Scholar] [CrossRef]
- Jayaraman, S. Flow cytometric determination of mitochondrial membrane potential changes during apoptosis of T lymphocytic and pancreatic beta cell lines: Comparison of tetramethylrhodamineethylester (TMRE), chloromethyl-X-rosamine (H2-CMX-Ros) and MitoTracker Red 580 (MTR580). J. Immunol. Methods 2005, 306, 68–79. [Google Scholar] [CrossRef]
- Schreer, A.; Tinson, C.; Sherry, J.; Schirmer, K. Application of Alamar blue/5-carboxyfluorescein diacetate acetoxymethyl ester as a noninvasive cell viability assay in primary hepatocytes from rainbow trout. Anal. Biochem. 2005, 344, 76–85. [Google Scholar] [CrossRef]
- Kim, S.G. Quantification of relative cerebral blood flow change by flow-sensitive alternating inversion recovery (FAIR) technique: Application to functional mapping. Magn. Reson. Med. 1995, 34, 293–301. [Google Scholar] [CrossRef]
- Bradley, W.G., Jr. MR appearance of hemorrhage in the brain. Radiology 1993, 189, 15–26. [Google Scholar] [CrossRef]
- Kaluz, S.; Kaluzova, M.; Chrastina, A.; Olive, P.L.; Pastorekova, S.; Pastorek, J.; Lerman, M.I.; Stanbridge, E.J. Lowered oxygen tension induces expression of the hypoxia marker MN/carbonic anhydrase IX in the absence of hypoxia-inducible factor 1 α stabilization: A role for phosphatidylinositol 3’-kinase. Cancer Res. 2002, 62, 4469–4477. [Google Scholar]
- Airley, R.E.; Loncaster, J.; Raleigh, J.A.; Harris, A.L.; Davidson, S.E.; Hunter, R.D.; West, C.M.; Stratford, I.J. GLUT-1 and CAIX as intrinsic markers of hypoxia in carcinoma of the cervix: Relationship to pimonidazole binding. Int. J. Cancer. J. Int. Du Cancer 2003, 104, 85–91. [Google Scholar] [CrossRef]
- Kaluz, S.; Kaluzova, M.; Stanbridge, E.J. Expression of the hypoxia marker carbonic anhydrase IX is critically dependent on SP1 activity. Identification of a novel type of hypoxia-responsive enhancer. Cancer Res. 2003, 63, 917–922. [Google Scholar]
- Jiang, F.; Zhang, X.; Kalkanis, S.N.; Zhang, Z.; Yang, H.; Katakowski, M.; Hong, X.; Zheng, X.; Zhu, Z.; Chopp, M. Combination therapy with antiangiogenic treatment and photodynamic therapy for the nude mouse bearing U87 glioblastoma. Photochem. Photobiol. 2008, 84, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Hebeda, K.M.; Kamphorst, W.; Sterenborg, H.J.; Wolbers, J.G. Damage to tumour and brain by interstitial photodynamic therapy in the 9L rat tumour model comparing intravenous and intratumoral administration of the photosensitiser. Acta Neurochir. 1998, 140, 495–501. [Google Scholar] [CrossRef] [PubMed]
- TYKERB® (lapatinib) [Package Insert]; U.S. Food and Drug Administration; Novartis Pharmaceuticals Corporation: East Hanover, NJ, USA, 2018.
- Moy, B.; Goss, P.E. Lapatinib-associated toxicity and practical management recommendations. Oncologist 2007, 12, 756–765. [Google Scholar] [CrossRef]
- Tangutoori, S.; Ohta, A.; Gatley, S.; Campbell, R.B. Repurposing an Erstwhile Cancer Drug: A Quantitative and Therapeutic Evaluation of Alternative Nanosystems for the Delivery of Colchicine to Solid Tumors. J. Cancer Sci. Ther. 2014, 6, 236–246. [Google Scholar] [CrossRef]
- Campbell, R.B.; Fukumura, D.; Brown, E.B.; Mazzola, L.M.; Izumi, Y.; Jain, R.K.; Torchilin, V.P.; Munn, L.L. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002, 62, 6831–6836. [Google Scholar] [PubMed]
- Friend, D.S.; Papahadjopoulos, D.; Debs, R.J. Endocytosis and intracellular processing accompanying transfection mediated by cationic liposomes. Biochim. Biophys. Acta 1996, 1278, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Romoren, K.; Thu, B.J.; Bols, N.C.; Evensen, O. Transfection efficiency and cytotoxicity of cationic liposomes in salmonid cell lines of hepatocyte and macrophage origin. Biochim. Biophys. Acta 2004, 1663, 127–134. [Google Scholar] [CrossRef]
- Rizvi, I.; Obaid, G.; Bano, S.; Hasan, T.; Kessel, D. Photodynamic therapy: Promoting in vitro efficacy of photodynamic therapy by liposomal formulations of a photosensitizing agent. Lasers Surg. Med. 2018, 50, 499–505. [Google Scholar] [CrossRef]
- Dai, C.L.; Tiwari, A.K.; Wu, C.P.; Su, X.D.; Wang, S.R.; Liu, D.G.; Ashby, C.R., Jr.; Huang, Y.; Robey, R.W.; Liang, Y.J.; et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008, 68, 7905–7914. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chen, Z.; Chen, L.; Wang, F.; Li, F.; Wang, X.; Fu, L. ABCG2-overexpressing H460/MX20 cell xenografts in athymic nude mice maintained original biochemical and cytological characteristics. Sci. Rep. 2017, 7, 40064. [Google Scholar] [CrossRef] [Green Version]
- Diaz, R.; Nguewa, P.A.; Parrondo, R.; Perez-Stable, C.; Manrique, I.; Redrado, M.; Catena, R.; Collantes, M.; Penuelas, I.; Diaz-Gonzalez, J.A.; et al. Antitumor and antiangiogenic effect of the dual EGFR and HER-2 tyrosine kinase inhibitor lapatinib in a lung cancer model. BMC Cancer 2010, 10, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaya, M.K.; Demir, T.; Bulut, H.; Akpolat, N.; Turgut, B. Effects of lapatinib and trastuzumab on vascular endothelial growth factor in experimental corneal neovascularization. Clin. Exp. Ophthalmol. 2015, 43, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Liu, L.; Wang, M.; Ding, K.; Fan, Y.; Ma, B.; Lal, B.; Tyler, B.; Mangraviti, A.; Wang, S.; et al. Quantitative multiparametric MRI assessment of glioma response to radiotherapy in a rat model. Neuro Oncol. 2014, 16, 856–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, A.; Loomis, K.; Smith, B.; Lee, J.H.; Yavlovich, A.; Heldman, E.; Blumenthal, R. Lipid-based nanoparticles as pharmaceutical drug carriers: From concepts to clinic. Crit. Rev. Drug Carr. Syst. 2009, 26, 523–580. [Google Scholar] [CrossRef] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisher, C.; Obaid, G.; Niu, C.; Foltz, W.; Goldstein, A.; Hasan, T.; Lilge, L. Liposomal Lapatinib in Combination with Low-Dose Photodynamic Therapy for the Treatment of Glioma. J. Clin. Med. 2019, 8, 2214. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8122214
Fisher C, Obaid G, Niu C, Foltz W, Goldstein A, Hasan T, Lilge L. Liposomal Lapatinib in Combination with Low-Dose Photodynamic Therapy for the Treatment of Glioma. Journal of Clinical Medicine. 2019; 8(12):2214. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8122214
Chicago/Turabian StyleFisher, Carl, Girgis Obaid, Carolyn Niu, Warren Foltz, Alyssa Goldstein, Tayyaba Hasan, and Lothar Lilge. 2019. "Liposomal Lapatinib in Combination with Low-Dose Photodynamic Therapy for the Treatment of Glioma" Journal of Clinical Medicine 8, no. 12: 2214. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8122214