Why Have Detection, Understanding and Management of Kidney Hypoxic Injury Lagged behind Those for the Heart?


Abstract
:1. Introduction
2. Comparing the Pathogenesis of Renal and Cardiac Hypoxic Injury
3. Conceptual Errors in the Understanding of Hypoxic AKI
4. Clinical Presentation of Hypoxic Cardiac vs. Renal Injury
5. Detection and Assessment of Myocardial Hypoxic Injury
6. Detection of Hypoxic Renal Injury
7. Biomarkers of Acute Kidney Injury
8. Renal Functional Imaging
9. Future Challenges
10. Conclusions
Author Contributions
Conflicts of Interest
References
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W.; Wang, W.; Poole, B.; Mitra, A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J. Clin. Investig. 2004, 114, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Moran, S.M.; Myers, B.D. Course of acute renal failure studied by a model of creatinine kinetics. Kidney Int. 1985, 27, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Smilowitz, N.R.; Naoulou, B.; Sedlis, S.P. Diagnosis and Management of Type II Myocardial Infarction: Increased Demand for a Limited Supply of Evidence. Curr. Atheroscler. Rep. 2015, 17, 478. [Google Scholar] [CrossRef] [PubMed]
- Bache, R.J.; Dai, X.Z. Myocardial oxygen consumption during exercise in the presence of left ventricular hypertrophy secondary to supravalvular aortic stenosis. J. Am. Coll. Cardiol. 1990, 15, 1157–1164. [Google Scholar] [CrossRef]
- Watanabe, H.; Ohtsuka, S.; Kakihana, M.; Sugishita, Y. Decreased aortic compliance aggravates subendocardial ischaemia in dogs with stenosed coronary artery. Cardiovasc. Res. 1992, 26, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Nagao, N.; Yamasaki, Y.; Kawanami, S.; Kamitani, T.; Sagiyama, K.; Higo, T.; Ide, T.; Takemura, A.; Ishizaki, U.; Fukushima, K.; et al. Quantification of myocardial oxygenation in heart failure using blood-oxygen-level-dependent T2* magnetic resonance imaging: Comparison with cardiopulmonary exercise test. Magn. Reson. Imaging 2017, 39, 138–143. [Google Scholar] [CrossRef] [PubMed]
- May, D.; Gilon, D.; Djonov, V.; Itin, A.; Lazarus, A.; Gordon, O.; Rosenberger, C.; Keshet, E. Transgenic system for conditional induction and rescue of chronic myocardial hibernation provides insights into genomic programs of hibernation. Proc. Natl. Acad. Sci. USA 2008, 105, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef]
- Buja, L.M. Myocardial ischemia and reperfusion injury. Cardiovasc. Pathol. 2005, 14, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Hazanov, N.; Somin, M.; Attali, M.; Beilinson, N.; Thaler, M.; Mouallem, M.; Maor, Y.; Zaks, N.; Malnick, S. Acute renal embolism: Forty-four cases of renal infarction in patients with atrial fibrillation. Medicine (Baltimore) 2004, 83, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Matlin, R.A.; Gary, N.E. Acute cortical necrosis. Case report and review of the literature. Am. J. Med. 1974, 56, 110–118. [Google Scholar] [CrossRef]
- Rosen, S.; Stillman, I.E. Acute Tubular Necrosis Is a Syndrome of Physiologic and Pathologic Dissociation. J. Am. Soc. Nephrol. 2008, 19, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Brezis, M.; Rosen, S. Hypoxia of the Renal Medulla-Its Implications for Disease. N. Engl. J. Med. 1995, 332, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Evans, R.G.; Rosen, S.; Rosenberger, C. Cellular adaptive changes in AKI: mitigating renal hypoxic injury. Nephrol. Dial. Transplant. 2012, 27, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Stillman, I.E.; Brezis, M.; Heyman, S.N.; Epstein, F.H.; Spokes, K.; Rosen, S. Effects of salt depletion on the kidney: changes in medullary oxygenation and thick ascending limb size. J. Am. Soc. Nephrol. 1994, 4, 1538–1545. [Google Scholar] [PubMed]
- Brezis, M.; Heyman, S.N.; Epstein, F.H. Determinants of intrarenal oxygenation. II. Hemodynamic effects. Am. J. Physiol. 1994, 267, F1063–F1068. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, N.; Rosenberger, C.; Heyman, S.N.; Eckardt, K.U. Regulation of hypoxia-inducible factor in kidney disease. Clin. Exp. Pharmacol. Physiol. 2013, 40, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Khamaisi, M.; Rosen, S.; Rosenberger, C. Renal parenchymal hypoxia, hypoxia response and the progression of chronic kidney disease. Am. J. Nephrol. 2008, 28, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Franzén, S.; Palm, F. Endothelin type A receptor inhibition normalises intrarenal hypoxia in rats used as a model of type 1 diabetes by improving oxygen delivery. Diabetologia 2015, 58, 2435–2442. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Rosenberger, C.; Rosen, S.; Khamaisi, M. Why is diabetes mellitus a risk factor for contrast-induced nephropathy? BioMed Res. Int. 2013, 2013, 123589. [Google Scholar] [CrossRef] [PubMed]
- Washington, J.A., 2nd; Holland, J.M. Urine oxygen tension: effects of osmotic and saline diuresis and of ethacrynic acid. Am. J. Physiol. 1966, 210, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Brezis, M.; Agmon, Y.; Epstein, F.H. Determinants of intrarenal oxygenation. I. Effects of diuretics. Am. J. Physiol. 1994, 267, F1063–F1068. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J.; Fasching, A.; Pihl, L.; Patinha, D.; Franzén, S.; Palm, F. Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol. Renal Physiol. 2015, 309, F227–F234. [Google Scholar] [CrossRef] [PubMed]
- Szalat, A.; Perlman, A.; Muszkat, M.; Khamaisi, M.; Abassi, Z.; Heyman, S.N. Can SGLT2 Inhibitors Cause Acute Renal Failure? Plausible Role for Altered Glomerular Hemodynamics and Medullary Hypoxia. Drug Saf. 2018, 41, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.; Rosen, S.; Heyman, S.N. Renal parenchymal oxygenation and hypoxia adaptation in acute kidney injury. Clin. Exp. Pharmacol. Physiol. 2006, 33, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Rosen, S.; Rosenberger, C. Renal parenchymal hypoxia, hypoxia adaptation, and the pathogenesis of radiocontrast nephropathy. Clin. J. Am. Soc. Nephrol. 2008, 3, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Gorelik, Y.; Zorbavel, D.; Rosenberger, C.; Abassi, Z.; Rosen, S. Near drowning: New perspectives for human hypoxic AKI. Nephrol Dial. Transplant. 2019. [Google Scholar] [CrossRef] [PubMed]
- Brezis, M.; Shanley, P.; Silva, P.; Spokes, K.; Lear, S.; Epstein, F.H.; Rosen, S. Disparate mechanisms for hypoxic cell injury in different nephron segments. Studies in the isolated perfused rat kidney. J. Clin. Investig. 1985, 76, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Brezis, M.; Rosen, S.; Silva, P.; Epstein, F.H. Transport activity modifies thick ascending limb damage in the isolated perfused kidney. Kidney Int. 1984, 25, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Shanley, P.F.; Brezis, M.; Spokes, K.; Silva, P.; Epstein, F.H.; Rosen, S. Transport-dependent cell injury in the S3 segment of the proximal tubule. Kidney Int. 1986, 29, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.; Rosen, S.; Shina, A.; Bernhardt, W.; Wiesener, M.S.; Frei, U.; Eckardt, K.U.; Heyman, S.N. Hypoxia-inducible factors and tubular cell survival in isolated perfused kidneys. Kidney Int. 2006, 70, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Rosen, S.; Epstein, F.H.; Spokes, K.; Brezis, M.L. Loop diuretics reduce hypoxic damage to proximal tubules of the isolated perfused rat kidney. Kidney Int. 1994, 45, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Shanley, P.F.; Rosen, M.D.; Brezis, M.; Silva, P.; Epstein, F.H.; Rosen, S. Topography of focal proximal tubular necrosis after ischemia with reflow in the rat kidney. Am. J. Pathol. 1985, 122, 462–468. [Google Scholar]
- Heyman, S.N. Proximal tubular injury attenuates outer medullary hypoxic damage: studies in perfused rat kidneys. Exp. Nephrol. 2001, 10, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Rosen, S.; Rosenberger, C. Critical assessment of experimental models of acute renal failure. Crit. Care Nephrol. 2009, 169, 237–250. [Google Scholar]
- Heyman, S.N.; Rosen, S.; Rosenberger, C. Animal models of renal dysfunction: acute kidney injury. Expert Opin. Drug Discov. 2009, 4, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Brezis, M.; Epstein, F.H.; Spokes, K.; Silva, P.; Rosen, S. Early renal medullary hypoxic injury from radiocontrast and indomethacin. Kidney Int. 1991, 40, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Brezis, M.; Reubinoff, C.A.; Greenfeld, Z.; Lechene, G.; Epstein, F.H.; Rosen, S. Acute renal failure with selective medullary injury in the rat. J. Clin. Investig. 1988, 82, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Rosenberger, C.; Rosen, S. Experimental ischemia-reperfusion: Biases and mythsthe proximal vs. distal hypoxic tubular injury debate revisited. Kidney Int. 2010, 77, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, R.; Ronco, C.; Kellum, J.A.; Mehta, R.L.; Palevsky, P.; Acute Dialysis Quality Initiative Workgroup. Acute renal failure-definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit. Care 2004, 8, R204–R212. [Google Scholar] [PubMed]
- Bagga, A.; Bakkaloglu, A.; Devarajan, P.; Mehta, R.L.; Kellum, J.A.; Shah, S.V.; Molitoris, B.A.; Ronco, C.; Warnock, D.G.; Joannidis, M.; et al. Improving outcomes from acute kidney injury: Report of an initiative. Pediatr. Nephrol. 2007, 22, 1655–1658. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C. Acute kidney injury: from clinical to molecular diagnosis. Crit. Care 2016, 20, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Cox, E.F.; Buchanan, C.E.; Bradley, C.R.; Prestwich, B.; Mahmoud, H.; Taal, M.; Selby, N.M.; Francis, S.T. Multiparametric renal magnetic resonance imaging: Validation, interventions, and alterations in chronic kidney disease. Front. Physiol. 2017, 8, 696. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, P. Review: Neutrophil gelatinase-associated lipocalin: A troponin-like biomarker for human acute kidney injury. Nephrology (Carlton) 2010, 15, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, V.S.; Ferguson, M.A.; Bonventre, J.V. Biomarkers of acute kidney injury: early diagnosis, pathogenesis, and recovery. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 463–493. [Google Scholar] [CrossRef] [PubMed]
- Vanmassenhove, J.; Vanholder, R.; Nagler, E.; Van Biesen, W. Urinary and serum biomarkers for the diagnosis of acute kidney injury: An in-depth review of the literature. Nephrol. Dial. Transplant. 2013, 28, 254–273. [Google Scholar] [CrossRef] [PubMed]
- Beker, B.M.; Corleto, M.G.; Fieiras, C.; Musso, C.G. Novel acute kidney injury biomarkers: Their characteristics, utility and concerns. Int. Urol. Nephrol. 2018, 50, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Qing, M.A.; Prada, A.; Mitsnefes, M.; Zahedi, K.; Yang, J.; Barasch, J.; Devarajan, P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J. Am. Soc. Nephrol. 2003, 14, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Dent, C.; Tarabishi, R.; Mitsnefes, M.M.; Ma, Q.; Kelly, C.; Ruff, S.M.; Zahedi, K.; Shao, M.; Bean, J.; et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 2005, 365, 1231–1238. [Google Scholar] [CrossRef]
- Haase, M.; Bellomo, R.; Devarajan, P.; Schlattmann, P.; Haase-Fielitz, A.; Bagshaw, S.M.; Bogle, R.; Changchun, C.; Constantin, J.M.; Cruz, D.; et al. Accuracy of Neutrophil Gelatinase-Associated Lipocalin (NGAL) in Diagnosis and Prognosis in Acute Kidney Injury: A Systematic Review and Meta-analysis. Am. J. Kidney Dis. 2009, 54, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, T.; Asseldonk, E.J.; Humphreys, B.D.; Gunaratnam, L.; Duffield, J.S.; Bonventre, J.V. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J. Clin. Investig. 2008, 118, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, T.; Bonventre, J.V.; Bailly, V.; Wei, H.; Hession, C.A.; Cate, R.L.; Sanicola, M. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J. Biol. Chem. 1998, 273, 4135–4142. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, V.Y.; Ecder, T.; Fantuzzi, G.; Siegmund, B.; Lucia, M.S.; Dinarello, C.A.; Schrier, R.W.; Edelstein, C.L. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J. Clin. Investig. 2001, 107, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.; Zhou, X. Comparison of urine neutrophil gelatinase-associated lipocalin and interleukin-18 in prediction of acute kidney injury in adults. Medicine (Baltimore) 2018, 97, e12570. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guo, W.; Zhang, J.; Xu, C.; Yu, S.; Mao, Z.; Wu, J.; Ye, C.; Mei, C.; Dai, B. Urinary interleukin 18 for detection of acute kidney injury: A meta-analysis. Am. J. Kidney Dis. 2013, 62, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.M.; Huang, L.F.; Zheng, Y.; Li, W.X. Diagnostic value of urinary tissue inhibitor of metalloproteinase-2 and insulin-like growth factor binding protein 7 for acute kidney injury: A meta-analysis. Crit. Care 2017, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Hemdahl, A.L.; Gabrielsen, A.; Zhu, C.; Eriksson, P.; Hedin, U.; Kastrup, J.; Thorén, P.; Hansson, G.K. Expression of neutrophil gelatinase-associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Elmedany, S.M.; Naga, S.S.; Elsharkawy, R.; Mahrous, R.S.; Elnaggar, A.I. Novel urinary biomarkers and the early detection of acute kidney injury after open cardiac surgeries. J. Crit. Care 2017, 40, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Ghatanatti, R.; Teli, A.; Tirkey, S.S.; Bhattacharya, S.; Sengupta, G.; Mondal, A. Role of renal biomarkers as predictors of acute kidney injury in cardiac surgery. Asian Cardiovasc. Thorac. Ann. 2014, 22, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Buddenkotte, J.; Steinhoff, M. Recent advances in understanding and managing. F1000 Res. 2018, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Petcharunpaisan, S. Arterial spin labeling in neuroimaging. World J. Radiol. 2010, 2, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Nery, F.; Gordon, I.; Thomas, D. Non-Invasive Renal Perfusion Imaging Using Arterial Spin Labeling MRI: Challenges and Opportunities. Diagnostics (Basel) 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Odudu, A.; Nery, F.; Harteveld, A.A.; Evans, R.G.; Pendse, D.; Buchanan, C.E.; Francis, S.T.; Fernández-Seara, M.A. Arterial spin labelling MRI to measure renal perfusion: a systematic review and statement paper. Nephrol. Dial. Transplant. 2018, 33, ii15–ii21. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, J.B.; Shaw, A.D.; Billings, F.T. Acute kidney injury following cardiac surgery: Current understanding and future directions. Crit. Care 2016, 20, 187. [Google Scholar] [CrossRef] [PubMed]
- Hueper, K.; Gueler, F.; Bräsen, J.H.; Gutberlet, M.; Jang, M.S.; Lehner, F.; Richter, N.; Hanke, N.; Peperhove, M.; Martirosian, P.; et al. Functional MRI detects perfusion impairment in renal allografts with delayed graft function. Am. J. Physiol. Ren. Physiol. 2015, 308, F1444–F1451. [Google Scholar] [CrossRef] [PubMed]
- Hueper, K.; Gutberlet, M.; Rong, S.; Hartung, D.; Mengel, M.; Lu, X.; Haller, H.; Wacker, F.; Meier, M.; Gueler, F. Acute Kidney Injury: Arterial Spin Labeling to Monitor Renal Perfusion Impairment in Mice-Comparison with Histopathologic Results and Renal Function. Radiology 2014, 270, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.V.; Priatna, A.; Spokes, K.; Epstein, F.H. Changes in intrarenal oxygenation as evaluated by BOLD MRI in a rat kidney model for radiocontrast nephropathy. J. Magn. Reson. Imaging 2001, 13, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Pruijm, M.; Mendichovszky, I.A.; Liss, P.; Van der Niepen, P.; Textor, S.C.; Lerman, L.O.; Krediet, C.T.P.; Caroli, A.; Burnier, M.; Prasad, P.V. Renal blood oxygenation level-dependent magnetic resonance imaging to measure renal tissue oxygenation: a statement paper and systematic review. Nephrol. Dial. Transplant. 2018, 33, ii22–ii28. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.V.; Edelman, R.R.; Epstein, F.H. Noninvasive evaluation of intrarenal oxygenation with BOLD MRI. Circulation 1996, 94, 3271–3275. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.V.; Chen, Q.; Goldfarb, J.W.; Epstein, F.H.; Edelman, R.R. Breath-hold R2 mapping with a multiple gradient-recalled echo sequence: Application to the evaluation of intrarenal oxygenation. J. Magn. Reson. Imaging 1997, 7, 1163–1165. [Google Scholar] [CrossRef] [PubMed]
- Textor, S.C.; Glockner, J.F.; Lerman, L.O.; Misra, S.; McKusick, M.A.; Riederer, S.J.; Grande, J.P.; Gomez, S.I.; Romero, J.C. The Use of Magnetic Resonance to Evaluate Tissue Oxygenation in Renal Artery Stenosis. J. Am. Soc. Nephrol. 2008, 19, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Morrell, G.; Rusinek, H.; Sigmund, E.E.; Chandarana, H.; Lerman, L.O.; Prasad, P.V.; Niles, D.; Artz, N.; Fain, S.; et al. New magnetic resonance imaging methods in nephrology. Kidney Int. 2014, 85, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Bokacheva, L.; Rusinek, H.; Chen, Q.; Oesingmann, N.; Prince, C.; Kaur, M.; Kramer, E.; Lee, V.S. Quantitative determination of Gd-DTPA concentration in T1-weighted MR renography studies. Magn. Reson. Med. 2007, 57, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Bokacheva, L.; Rusinek, H.; Zhang, J.L.; Chen, Q.; Lee, V.S. Estimates of glomerular filtration rate from MR renography and tracer kinetic models. J. Magn. Reson. Imaging 2009, 29, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Milman, Z.; Heyman, S.N.; Corchia, N.; Edrei, Y.; Axelrod, J.H.; Rosenberger, C.; Tsarfati, G.; Abramovitch, R. Hemodynamic response magnetic resonance imaging: Application for renal hemodynamic characterization. Nephrol. Dial. Transplant. 2013, 28, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Milman, Z.; Axelrod, J.H.; Heyman, S.N.; Nachmansson, N.; Abramovitch, R. Assessment with unenhanced MRI techniques of renal morphology and hemodynamic changes during acute kidney injury and chronic kidney disease in mice. Am. J. Nephrol. 2014, 39, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Maril, N.; Margalit, R.; Rosen, S.; Heyman, S.N.; Degani, H. Detection of evolving acute tubular necrosis with renal 23Na MRI: Studies in rats. Kidney Int. 2006, 69, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Kleeman, C.R.; Maxwell, M.H.; Witlin, S. Functional isosthenuria; an isolated reversible renal tubular defect. AMA Arch. Intern. Med. 1958, 101, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.Z.L.; Martin, A.; Cochrane, A.D.; Smith, J.A.; Thrift, A.G.; Harrop, G.K.; Ngo, J.P.; Evans, R.G. Urinary hypoxia: an intraoperative marker of risk of cardiac surgery-associated acute kidney injury. Nephrol Dial Transplant. 2018, 33, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Reshef, A.; Shirvan, A.; Akselrod-Ballin, A.; Wall, A.; Ziv, I. Small-Molecule Biomarkers for Clinical PET Imaging of Apoptosis. J. Nucl. Med. 2010, 51, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Damianovich, M.; Ziv, I.; Heyman, S.N.; Rosen, S.; Shina, A.; Kidron, D.; Aloya, T.; Grimberg, H.; Levin, G.; Reshef, A.; et al. ApoSense: A novel technology for functional molecular imaging of cell death in models of acute renal tubular necrosis. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Correas, J.M.; Anglicheau, D.; Joly, D.; Gennisson, J.L.; Tanter, M.; Hélénon, O. Ultrasound-based imaging methods of the kidney-recent developments. Kidney Int. 2016, 90, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Solomon, R.; Werner, C.; Mann, D.; D’Elia, J.; Silva, P. Effects of Saline, Mannitol, and Furosemide on Acute Decreases in Renal Function Induced by Radiocontrast Agents. N. Engl. J. Med. 1994, 331, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.M.; Heyman, S.; Brezis, M. Potential deleterious effect of furosemide in radiocontrast nephropathy. Nephron 1992, 62, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Brezis, M.; Greenfeld, Z.; Rosen, S. Protective Role of Furosemide and Saline in Radiocontrast-Induced Acute Renal Failure in the Rat. Am. J. Kidney Dis. 1989, 14, 377–385. [Google Scholar] [CrossRef]
- Briguori, C.; Visconti, G.; Focaccio, A.; Airoldi, F.; Valgimigli, M.; Sangiorgi, G.M.; Golia, B.; Ricciardelli, B.; Condorelli, G. Renal Insufficiency After Contrast Media Administration Trial II (REMEDIAL II). Circulation 2011, 124, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, M.; Rosenberger, C.; Abassi, Z.; Shina, A.; Zilbersat, F.; Eckardt, K.U.; Rosen, S.; Heyman, S.N. Acute-on-chronic renal failure in the rat: Functional compensation and hypoxia tolerance. Am. J. Nephrol. 2006, 26, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.; Heyman, S.N.; Rosen, S.; Shina, A.; Goldfarb, M.; Griethe, W.; Frei, U.; Reinke, P.; Bachmann, S.; Eckardt, K.U. Up-regulation of HIF in experimental acute renal failure: Evidence for a protective transcriptional response to hypoxia. Kidney Int. 2005, 67, 531–542. [Google Scholar] [CrossRef] [PubMed]



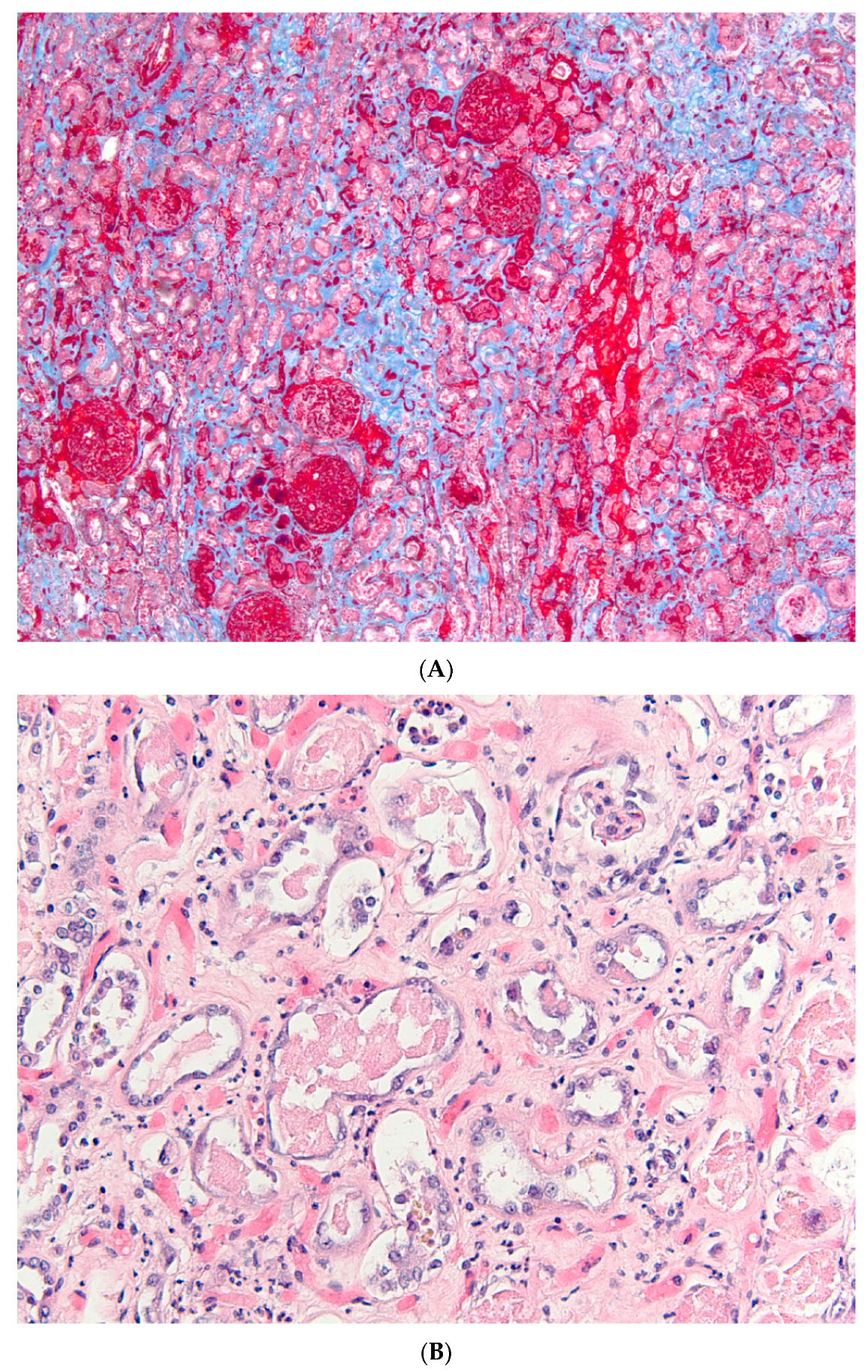{kind=link}
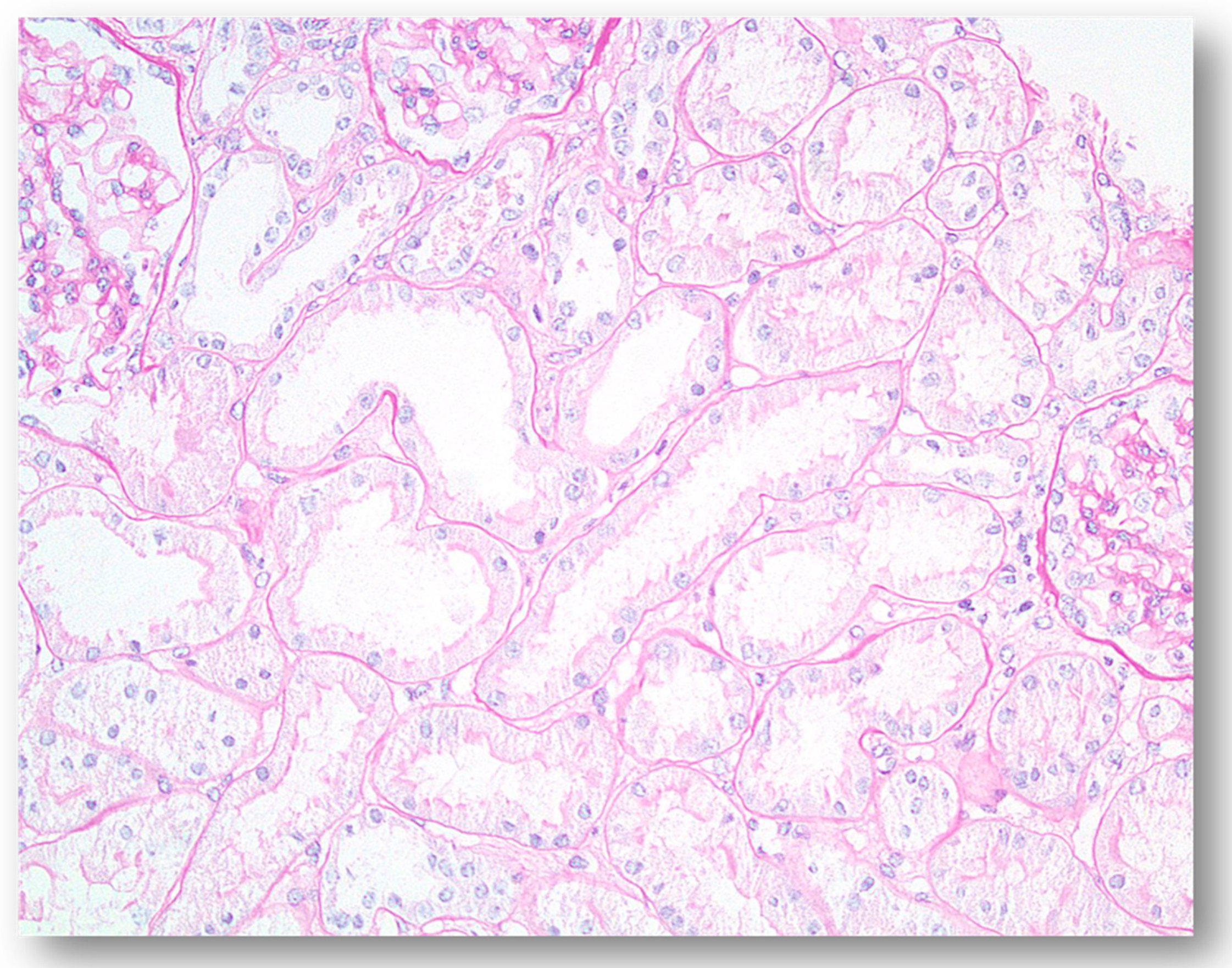{kind=link}
| Type I Ischemic AKI | Type II Hypoxic AKI | Type I AMI | Type II AMI | |
|---|---|---|---|---|
| Pathophysiology | Ischemia and reperfusion | Altered oxygen supply/demand balance | Ischemia and reperfusion | Altered oxygen supply/demand balance |
| Tissue blood supply | ceases | Maintained or reduced | ceases | Maintained or reduced |
| Workload and oxygen consumption | ceases | continued/enhanced | Rapidly declines | enhanced |
| Clinical scenario | Rare Renal infarct | Common Hypotension, CKD, diabetes, NSAIDs, iodinated contrast media, osmotic dieresis etc. | AMI Due to ruptured plaque | Increased workload in the presence of coronary stenosis (hypotension, anemia, tachycardia etc.) |
| Symptoms | Flank pain, hematuria | None related to AKI | Chest pain and accompanying complaints | Chest pain and accompanying complaints |
| Immediate evidence of functional impairment | None if unilateral/segmental | None | Occasionally evidence of impaired circulation and heart failure | Occasionally evidence of impaired circulation and heart failure |
| Rapid functional diagnostic tools | None | None | Echocardiography (ECG) | Echocardiography (ECG) |
| Specific biomarkers | Available and sensitive, limited specificity † | Available, limited specificity and sensitivity † | Highly specific and sensitive in detecting injury | Highly specific and sensitive in detecting injury |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abassi, Z.; Rosen, S.; Lamothe, S.; Heyman, S.N. Why Have Detection, Understanding and Management of Kidney Hypoxic Injury Lagged behind Those for the Heart? J. Clin. Med. 2019, 8, 267. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8020267
Abassi Z, Rosen S, Lamothe S, Heyman SN. Why Have Detection, Understanding and Management of Kidney Hypoxic Injury Lagged behind Those for the Heart? Journal of Clinical Medicine. 2019; 8(2):267. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8020267
Chicago/Turabian StyleAbassi, Zaid, Seymour Rosen, Simon Lamothe, and Samuel N. Heyman. 2019. "Why Have Detection, Understanding and Management of Kidney Hypoxic Injury Lagged behind Those for the Heart?" Journal of Clinical Medicine 8, no. 2: 267. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8020267