Consequences of EMT-Driven Changes in the Immune Microenvironment of Breast Cancer and Therapeutic Response of Cancer Cells

Abstract
:1. Introduction
2. Breast Cancer Subtypes and Their Association with EMT
2.1. Luminal A and B Breast Cancers and EMT
2.2. HER2-Positive Breast Cancer and EMT
2.3. TNBC or Basal-Like, Claudin-Low Breast Cancer and EMT
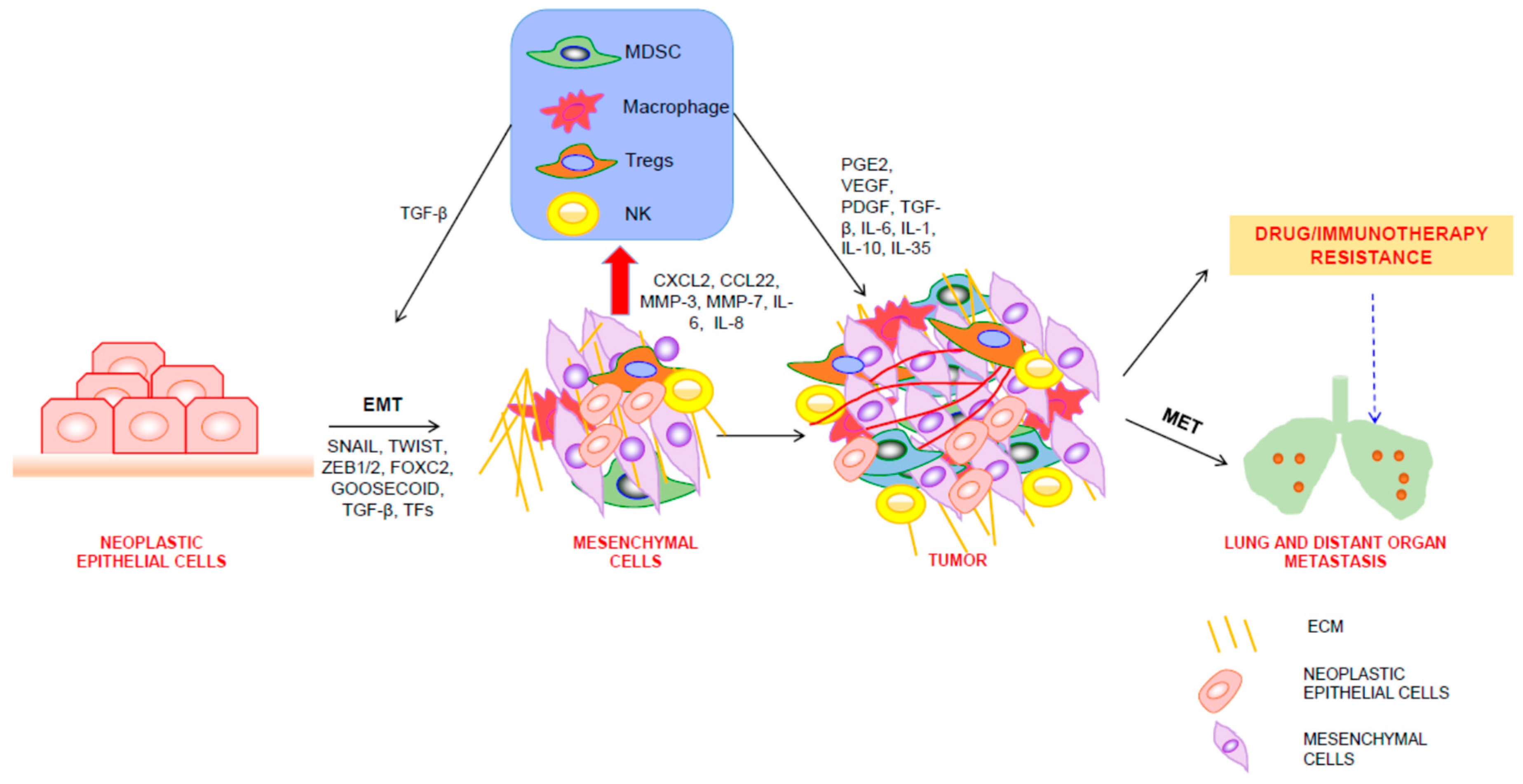
3. EMT Shapes the TME
4. Regulation of EMT by Immune Cells in TME
5. EMT, TME, and Therapeutic Resistance of Tumor Cells
5.1. Chemotherapy
5.2. Immunotherapy
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Desmedt, C.; Haibe-Kains, B.; Wirapati, P.; Buyse, M.; Larsimont, D.; Bontempi, G.; Delorenzi, M.; Piccart, M.; Sotiriou, C. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin. Cancer Res. 2008, 14, 5158–5165. [Google Scholar] [CrossRef]
- Rouzier, R.; Perou, C.M.; Symmans, W.F.; Ibrahim, N.; Cristofanilli, M.; Anderson, K.; Hess, K.R.; Stec, J.; Ayers, M.; Wagner, P.; et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin. Cancer Res. 2005, 11, 5678–5685. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Del Pozo Martin, Y.; Park, D.; Ramachandran, A.; Ombrato, L.; Calvo, F.; Chakravarty, P.; Spencer-Dene, B.; Derzsi, S.; Hill, C.S.; Sahai, E.; et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation. Epithelial Reversion, and Metastatic Colonization. Cell Rep. 2015, 13, 2456–2469. [Google Scholar] [CrossRef] [PubMed]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massague, J.; Benezra, R. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Brenot, A.; Knolhoff, B.L.; DeNardo, D.G.; Longmore, G.D. SNAIL1 action in tumor cells influences macrophage polarization and metastasis in breast cancer through altered GM-CSF secretion. Oncogenesis 2018, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Cortes, M.; Sanchez-Moral, L.; de Barrios, O.; Fernandez-Acenero, M.J.; Martinez-Campanario, M.C.; Esteve-Codina, A.; Darling, D.S.; Gyorffy, B.; Lawrence, T.; Dean, D.C.; et al. Tumor-associated macrophages (TAMs) depend on ZEB1 for their cancer-promoting roles. EMBO J. 2017, 36, 3336–3355. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Iida, N.; Roberts, E.W.; Sangaletti, S.; Wong, M.H.; Yull, F.E.; Coussens, L.M.; DeClerck, Y.A. Tumor microenvironment complexity: Emerging roles in cancer therapy. Cancer Res. 2012, 72, 2473–2480. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1. PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [Green Version]
- Yerushalmi, R.; Woods, R.; Ravdin, P.M.; Hayes, M.M.; Gelmon, K.A. Ki67 in breast cancer: Prognostic and predictive potential. Lancet Oncol. 2010, 11, 174–183. [Google Scholar] [CrossRef]
- Cheang, M.C.; Chia, S.K.; Voduc, D.; Gao, D.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 index. HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Agus, D.B.; Akita, R.W.; Fox, W.D.; Lewis, G.D.; Higgins, B.; Pisacane, P.I.; Lofgren, J.A.; Tindell, C.; Evans, D.P.; Maiese, K.; et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2002, 2, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Hung, M.C. Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene 2000, 19, 6115–6121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-like breast cancer: A critical review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef] [PubMed]
- Yehiely, F.; Moyano, J.V.; Evans, J.R.; Nielsen, T.O.; Cryns, V.L. Deconstructing the molecular portrait of basal-like breast cancer. Trends Mol. Med. 2006, 12, 537–544. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Gilcrease, M.Z.; Krishnamurthy, S.; Lee, J.S.; Fridlyand, J.; Sahin, A.; Agarwal, R.; Joy, C.; et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009, 69, 4116–4124. [Google Scholar] [CrossRef]
- Lindley, L.E.; Briegel, K.J. Molecular characterization of TGFbeta-induced epithelial-mesenchymal transition in normal finite lifespan human mammary epithelial cells. Biochem. Biophys. Res. Commun. 2010, 399, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Franklin, C.K.; Mackay, A.; Reis-Filho, J.S.; Isacke, C.M. Epithelial and mesenchymal subpopulations within normal basal breast cell lines exhibit distinct stem cell/progenitor properties. Stem Cells 2012, 30, 292–303. [Google Scholar] [CrossRef]
- Kang, Y.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell. 2008, 14, 818–829. [Google Scholar] [CrossRef]
- Braune, E.B.; Seshire, A.; Lendahl, U. Notch and Wnt Dysregulation and Its Relevance for Breast Cancer and Tumor Initiation. Biomedicines 2018, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Cleary, A.S.; Leonard, T.L.; Gestl, S.A.; Gunther, E.J. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 2014, 508, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Guan, H.; Fang, L.; Yang, Y.; Zhu, X.; Yuan, J.; Wu, J.; Li, M. MicroRNA-374a activates Wnt/beta-catenin signaling to promote breast cancer metastasis. J. Clin. Investig. 2013, 123, 566–579. [Google Scholar] [PubMed]
- Kumar, S.; Srivastav, R.K.; Wilkes, D.W.; Ross, T.; Kim, S.; Kowalski, J.; Chatla, S.; Zhang, Q.; Nayak, A.; Guha, M.; et al. Estrogen-dependent DLL1-mediated Notch signaling promotes luminal breast cancer. Oncogene 2019, 38, 2092–2107. [Google Scholar] [CrossRef] [PubMed]
- Yook, J.I.; Li, X.Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J.; et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Lv, J.; Li, A.; Qiao, J.; Fang, L.; Li, Z.; Li, B.; Zhao, W.; Chen, H.; Wang, L. Constitutive expression of Bcl-2 induces epithelial-Mesenchymal transition in mammary epithelial cells. BMC Cancer 2015, 15, 476. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Pusztai, L. Gene expression profiling in breast cancer: Classification. prognostication, and prediction. Lancet 2011, 378, 1812–1823. [Google Scholar] [CrossRef]
- Ye, Y.; Xiao, Y.; Wang, W.; Yearsley, K.; Gao, J.X.; Shetuni, B.; Barsky, S.H. ERalpha signaling through slug regulates E-cadherin and EMT. Oncogene 2010, 29, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Belguise, K.; Kersual, N.; Kirsch, K.H.; Mineva, N.D.; Galtier, F.; Chalbos, D.; Sonenshein, G.E. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 2007, 9, 470–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.L.; Dou, X.W.; Bai, J.W.; Luo, X.R.; Qiu, S.Q.; Xi, D.D.; Huang, W.H.; Du, C.W.; Man, K.; Zhang, G.J. ERalpha inhibits epithelial-mesenchymal transition by suppressing Bmi1 in breast cancer. Oncotarget 2015, 6, 21704–21717. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Lehmann, W.; Mossmann, D.; Kleemann, J.; Mock, K.; Meisinger, C.; Brummer, T.; Herr, R.; Brabletz, S.; Stemmler, M.P.; Brabletz, T. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat. Commun. 2016, 7, 10498. [Google Scholar] [CrossRef] [Green Version]
- Pang, M.F.; Georgoudaki, A.M.; Lambut, L.; Johansson, J.; Tabor, V.; Hagikura, K.; Jin, Y.; Jansson, M.; Alexander, J.S.; Nelson, C.M.; et al. TGF-beta1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene 2016, 35, 748–760. [Google Scholar] [CrossRef]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef]
- Tian, M.; Schiemann, W.P. TGF-beta Stimulation of EMT Programs Elicits Non-genomic ER-alpha Activity and Anti-estrogen Resistance in Breast Cancer Cells. J. Cancer Metastasis Treat. 2017, 3, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grunert, S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: Dissection of Ras signaling pathways. J. Cell Biol. 2002, 156, 299–313. [Google Scholar] [CrossRef]
- Ning, Q.; Liu, C.; Hou, L.; Meng, M.; Zhang, X.; Luo, M.; Shao, S.; Zuo, X.; Zhao, X. Vascular endothelial growth factor receptor-1 activation promotes migration and invasion of breast cancer cells through epithelial-mesenchymal transition. PLoS ONE 2013, 8, e65217. [Google Scholar] [CrossRef] [PubMed]
- Chihara, Y.; Shimoda, M.; Hori, A.; Ohara, A.; Naoi, Y.; Ikeda, J.I.; Kagara, N.; Tanei, T.; Shimomura, A.; Shimazu, K.; et al. A small-molecule inhibitor of SMAD3 attenuates resistance to anti-HER2 drugs in HER2-positive breast cancer cells. Breast Cancer Res. Treat. 2017, 166, 55–68. [Google Scholar] [CrossRef]
- Goyette, M.A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018, 23, 1476–1490. [Google Scholar] [CrossRef]
- Gupta, P.; Srivastava, S.K. HER2 mediated de novo production of TGFbeta leads to SNAIL driven epithelial-to-mesenchymal transition and metastasis of breast cancer. Mol. Oncol. 2014, 8, 1532–1547. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Shi, Q.; Li, Y.; Xu, L.; Xu, C.; Chen, F.; Wang, H.; Liao, H.; Chang, Z.; Liu, F.; et al. HER2/EGFR-AKT Signaling Switches TGFbeta from Inhibiting Cell Proliferation to Promoting Cell Migration in Breast Cancer. Cancer Res. 2018, 78, 6073–6085. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Paw, I.; Dewhirst, M.W.; Lo, H.W. Akt phosphorylates and activates HSF-1 independent of heat shock, leading to Slug overexpression and epithelial-mesenchymal transition (EMT) of HER2-overexpressing breast cancer cells. Oncogene 2015, 34, 546–557. [Google Scholar] [CrossRef]
- Pomp, V.; Leo, C.; Mauracher, A.; Korol, D.; Guo, W.; Varga, Z. Differential expression of epithelial-mesenchymal transition and stem cell markers in intrinsic subtypes of breast cancer. Breast Cancer Res. Treat. 2015, 154, 45–55. [Google Scholar] [CrossRef]
- Hong, J.; Zhou, J.; Fu, J.; He, T.; Qin, J.; Wang, L.; Liao, L.; Xu, J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011, 71, 3980–3990. [Google Scholar] [CrossRef]
- Ai, M.; Liang, K.; Lu, Y.; Qiu, S.; Fan, Z. Brk/PTK6 cooperates with HER2 and Src in regulating breast cancer cell survival and epithelial-to-mesenchymal transition. Cancer Biol. Ther. 2013, 14, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.G.; Kim, S.J.; Kim, C.; Jeong, J. Molecular Classification of Triple-Negative Breast Cancer. J. Breast Cancer 2016, 19, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, Y.H.; Garcia-Garcia, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzman, M.; Grueso, J.; et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Chuang, H.C.; Kapuriya, N.; Kulp, S.K.; Chen, C.S.; Shapiro, C.L. Differential anti-proliferative activities of poly(ADP-ribose) polymerase (PARP) inhibitors in triple-negative breast cancer cells. Breast Cancer Res Treat. 2012, 134, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Anders, C.K.; Winer, E.P.; Ford, J.M.; Dent, R.; Silver, D.P.; Sledge, G.W.; Carey, L.A. Poly(ADP-Ribose) polymerase inhibition: “targeted” therapy for triple-negative breast cancer. Clin. Cancer Res. 2010, 16, 4702–4710. [Google Scholar] [CrossRef]
- Yoshida, T.; Ozawa, Y.; Kimura, T.; Sato, Y.; Kuznetsov, G.; Xu, S.; Uesugi, M.; Agoulnik, S.; Taylor, N.; Funahashi, Y.; et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br. J. Cancer 2014, 110, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Vikas, P.; Borcherding, N.; Zhang, W. The clinical promise of immunotherapy in triple-negative breast cancer. Cancer Manag. Res. 2018, 10, 6823–6833. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qi, Y.; Kong, X.; Zhai, J.; Li, Y.; Song, Y.; Wang, J.; Feng, X.; Fang, Y. Immunological therapy: A novel thriving area for triple-negative breast cancer treatment. Cancer Lett. 2019, 442, 409–428. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive. estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Sharma, A.; Balaji, S.A.; Gowda, M.C.; Dighe, R.R.; Kumar, R.V.; Rangarajan, A. Coordinate hyperactivation of Notch1 and Ras/MAPK pathways correlates with poor patient survival: Novel therapeutic strategy for aggressive breast cancers. Mol. Cancer Ther. 2014, 13, 3198–3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Shao, X.; Sun, H.; Liu, K.; Ding, Z.; Chen, J.; Fang, L.; Su, W.; Hong, Y.; Li, H.; et al. NUMB negatively regulates the epithelial-mesenchymal transition of triple-negative breast cancer by antagonizing Notch signaling. Oncotarget 2016, 7, 61036–61053. [Google Scholar] [CrossRef] [Green Version]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef] [Green Version]
- Choy, L.; Hagenbeek, T.J.; Solon, M.; French, D.; Finkle, D.; Shelton, A.; Venook, R.; Brauer, M.J.; Siebel, C.W. Constitutive NOTCH3 Signaling Promotes the Growth of Basal Breast Cancers. Cancer Res. 2017, 77, 1439–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speiser, J.; Foreman, K.; Drinka, E.; Godellas, C.; Perez, C.; Salhadar, A.; Ersahin, C.; Rajan, P. Notch-1 and Notch-4 biomarker expression in triple-negative breast cancer. Int. J. Surg. Pathol. 2012, 20, 139–145. [Google Scholar] [CrossRef]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of glioma-associated Oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFkappaB) pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef]
- Lei, J.; Fan, L.; Wei, G.; Chen, X.; Duan, W.; Xu, Q.; Sheng, W.; Wang, K.; Li, X. Gli-1 is crucial for hypoxia-induced epithelial-mesenchymal transition and invasion of breast cancer. Tumour Biol. 2015, 36, 3119–3126. [Google Scholar] [CrossRef] [PubMed]
- Hui, M.; Cazet, A.; Nair, R.; Watkins, D.N.; O’Toole, S.A.; Swarbrick, A. The Hedgehog signalling pathway in breast development. carcinogenesis and cancer therapy. Breast Cancer Res. 2013, 15, 203. [Google Scholar] [CrossRef] [PubMed]
- Woodward, W.A.; Chen, M.S.; Behbod, F.; Rosen, J.M. On mammary stem cells. J. Cell Sci. 2005, 118, 3585–3594. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhang, L.; He, X.; Zhang, P.; Sun, C.; Xu, X.; Lu, Y.; Li, F. TGF-beta plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem. Biophys. Res. Commun. 2018, 502, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Okita, Y.; Kimura, M.; Xie, R.; Chen, C.; Shen, L.T.; Kojima, Y.; Suzuki, H.; Muratani, M.; Saitoh, M.; Semba, K.; et al. The transcription factor MAFK induces EMT and malignant progression of triple-negative breast cancer cells through its target GPNMB. Sci. Signal. 2017, 10, eaak9397. [Google Scholar] [CrossRef] [PubMed]
- Granados-Principal, S.; Liu, Y.; Guevara, M.L.; Blanco, E.; Choi, D.S.; Qian, W.; Patel, T.; Rodriguez, A.A.; Cusimano, J.; Weiss, H.L.; et al. Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 2015, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Parvani, J.G.; Gujrati, M.D.; Mack, M.A.; Schiemann, W.P.; Lu, Z.R. Silencing beta3 Integrin by Targeted ECO/siRNA Nanoparticles Inhibits EMT and Metastasis of Triple-Negative Breast Cancer. Cancer Res. 2015, 75, 2316–2325. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Wei, Y.; Hwang, J.; Hang, X.; Andres Blanco, M.; Choudhury, A.; Tiede, B.; Romano, R.A.; DeCoste, C.; Mercatali, L.; et al. DeltaNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat. Cell Biol. 2014, 16, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar] [CrossRef] [PubMed]
- Vijay, G.V.; Zhao, N.; Den Hollander, P.; Toneff, M.J.; Joseph, R.; Pietila, M.; Taube, J.H.; Sarkar, T.R.; Ramirez-Pena, E.; Werden, S.J.; et al. GSK3beta regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 37. [Google Scholar] [CrossRef] [PubMed]
- El Ayachi, I.; Fatima, I.; Wend, P.; Alva-Ornelas, J.A.; Runke, S.; Kuenzinger, W.L.; Silva, J.; Silva, W.; Gray, J.K.; Lehr, S.; et al. The WNT10B Network Is Associated with Survival and Metastases in Chemoresistant Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 982–993. [Google Scholar] [CrossRef]
- Thiery, J.P.; Lim, C.T. Tumor dissemination: An EMT affair. Cancer Cell 2013, 23, 272–273. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef] [Green Version]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Chuang, L.S.; Ito, Y. RUNX3 is multifunctional in carcinogenesis of multiple solid tumors. Oncogene 2010, 29, 2605–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manandhar, S.; Lee, Y.M. Emerging role of RUNX3 in the regulation of tumor microenvironment. BMB Rep. 2018, 51, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Lin, J.H.; Brenot, A.; Kim, J.W.; Provot, S.; Werb, Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat. Cell Biol. 2013, 15, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Kouros-Mehr, H.; Bechis, S.K.; Slorach, E.M.; Littlepage, L.E.; Egeblad, M.; Ewald, A.J.; Pai, S.Y.; Ho, I.C.; Werb, Z. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell 2008, 13, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Tynan, J.A.; Wen, F.; Muller, W.J.; Oshima, R.G. Ets2-dependent microenvironmental support of mouse mammary tumors. Oncogene 2005, 24, 6870–6876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, A.K.; Young, L.J.; Tynan, J.A.; Lesperance, J.; Egeblad, M.; Werb, Z.; Hauser, C.A.; Muller, W.J.; Cardiff, R.D.; Oshima, R.G. Ets2-dependent stromal regulation of mouse mammary tumors. Mol. Cell Biol. 2003, 23, 8614–8625. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Hwang, J.; Andres Blanco, M.; Wei, Y.; Lukacisin, M.; Romano, R.A.; Smalley, K.; Liu, S.; Yang, Q.; Ibrahim, T.; et al. Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat. Cell Biol. 2012, 14, 1212–1222. [Google Scholar] [CrossRef]
- Kumar, S.; Wilkes, D.W.; Samuel, N.; Blanco, M.A.; Nayak, A.; Alicea-Torres, K.; Gluck, C.; Sinha, S.; Gabrilovich, D.; Chakrabarti, R. DeltaNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J. Clin. Investig. 2018, 128, 5095–5109. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D.; Silwal-Pandit, L.; Dannenfelser, R.; Langerod, A.; Vollan, H.K.; Vaske, C.; Siegel, J.U.; Troyanskaya, O.; Chin, S.F.; Caldas, C.; et al. Lymphocyte Invasion in IC10/Basal-Like Breast Tumors Is Associated with Wild-Type TP53. Mol. Cancer Res. 2015, 13, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef]
- Ha, H.Y.; Moon, H.B.; Nam, M.S.; Lee, J.W.; Ryoo, Z.Y.; Lee, T.H.; Lee, K.K.; So, B.J.; Sato, H.; Seiki, M.; et al. Overexpression of membrane-type matrix metalloproteinase-1 gene induces mammary gland abnormalities and adenocarcinoma in transgenic mice. Cancer Res. 2001, 61, 984–990. [Google Scholar]
- Sternlicht, M.D.; Lochter, A.; Sympson, C.J.; Huey, B.; Rougier, J.P.; Gray, J.W.; Pinkel, D.; Bissell, M.J.; Werb, Z. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 1999, 98, 137–146. [Google Scholar] [CrossRef]
- Masson, R.; Lefebvre, O.; Noel, A.; Fahime, M.E.; Chenard, M.P.; Wendling, C.; Kebers, F.; LeMeur, M.; Dierich, A.; Foidart, J.M.; et al. In vivo evidence that the stromelysin-3 metalloproteinase contributes in a paracrine manner to epithelial cell malignancy. J. Cell Biol. 1998, 140, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Wang, W.; Huang, X.R.; Oldfield, M.; Schmidt, A.M.; Cooper, M.E.; Lan, H.Y. Advanced glycation end products induce tubular epithelial-myofibroblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am. J. Pathol. 2004, 164, 1389–1397. [Google Scholar] [CrossRef]
- Petersen, O.W.; Nielsen, H.L.; Gudjonsson, T.; Villadsen, R.; Rank, F.; Niebuhr, E.; Bissell, M.J.; Ronnov-Jessen, L. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am. J. Pathol. 2003, 162, 391–402. [Google Scholar] [CrossRef]
- Del Casar, J.M.; Gonzalez, L.O.; Alvarez, E.; Junquera, S.; Marin, L.; Gonzalez, L.; Bongera, M.; Vazquez, J.; Vizoso, F.J. Comparative analysis and clinical value of the expression of metalloproteases and their inhibitors by intratumor stromal fibroblasts and those at the invasive front of breast carcinomas. Breast Cancer Res. Treat. 2009, 116, 39–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizoso, F.J.; Gonzalez, L.O.; Corte, M.D.; Rodriguez, J.C.; Vazquez, J.; Lamelas, M.L.; Junquera, S.; Merino, A.M.; Garcia-Muniz, J.L. Study of matrix metalloproteinases and their inhibitors in breast cancer. Br. J. Cancer 2007, 96, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Heppner, K.J.; Matrisian, L.M.; Jensen, R.A.; Rodgers, W.H. Expression of most matrix metalloproteinase family members in breast cancer represents a tumor-induced host response. Am. J. Pathol. 1996, 149, 273–282. [Google Scholar]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial Neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef]
- Lippitz, B.E. Cytokine patterns in patients with cancer: A systematic review. Lancet Oncol. 2013, 14, e218–e228. [Google Scholar] [CrossRef]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Chomarat, P.; Banchereau, J.; Davoust, J.; Palucka, A.K. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat. Immunol. 2000, 1, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Junius, S.; Benoy, I.; Van Dam, P.; Vermeulen, P.; Van Marck, E.; Huget, P.; Dirix, L.Y. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int. J. Cancer 2003, 103, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef]
- Xie, G.; Yao, Q.; Liu, Y.; Du, S.; Liu, A.; Guo, Z.; Sun, A.; Ruan, J.; Chen, L.; Ye, C.; et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int. J. Oncol. 2012, 40, 1171–1179. [Google Scholar]
- Liu, Q.; Yu, S.; Li, A.; Xu, H.; Han, X.; Wu, K. Targeting interlukin-6 to relieve immunosuppression in tumor microenvironment. Tumour Biol. 2017, 39, 1010428317712445. [Google Scholar] [CrossRef]
- Dominguez, C.; McCampbell, K.K.; David, J.M.; Palena, C. Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI Insight. 2017. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Yoshimura, T. The production of monocyte chemoattractant protein-1 (MCP-1)/CCL2 in tumor microenvironments. Cytokine 2017, 98, 71–78. [Google Scholar] [CrossRef]
- Ding, J.; Guo, C.; Hu, P.; Chen, J.; Liu, Q.; Wu, X.; Cao, Y.; Wu, J. CSF1 is involved in breast cancer progression through inducing monocyte differentiation and homing. Int. J. Oncol. 2016, 49, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration. invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Ostling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [Green Version]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Ma, B.; Shao, H.; Clark, A.M.; Wells, A. Macrophage phenotypic subtypes diametrically regulate epithelial-mesenchymal plasticity in breast cancer cells. BMC Cancer 2016, 16, 419. [Google Scholar] [CrossRef]
- Barbera-Guillem, E.; Nyhus, J.K.; Wolford, C.C.; Friece, C.R.; Sampsel, J.W. Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis. and IgG immune complexes potentiate the process. Cancer Res. 2002, 62, 7042–7049. [Google Scholar] [PubMed]
- Muraoka, R.S.; Dumont, N.; Ritter, C.A.; Dugger, T.C.; Brantley, D.M.; Chen, J.; Easterly, E.; Roebuck, L.R.; Ryan, S.; Gotwals, P.J.; et al. Blockade of TGF-beta inhibits mammary tumor cell viability. migration, and metastases. J. Clin. Investig. 2002, 109, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Bednarczyk, R.B.; Tuli, N.Y.; Hanly, E.K.; Rahoma, G.B.; Maniyar, R.; Mittelman, A.; Geliebter, J.; Tiwari, R.K. Macrophage inflammatory factors promote epithelial-mesenchymal transition in breast cancer. Oncotarget 2018, 9, 24272–24282. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Wang, X.H.; Gao, S.T.; Chen, C.; Xu, X.Y.; Sun, Q.; Zhou, Z.H.; Wu, G.Z.; Yu, Q.; Xu, G.; et al. Tumor-associated macrophages correlate with phenomenon of epithelial-mesenchymal transition and contribute to poor prognosis in triple-negative breast cancer patients. J. Surg. Res. 2018, 222, 93–101. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha. and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jochems, C.; Talaie, T.; Anderson, A.; Jales, A.; Tsang, K.Y.; Madan, R.A.; Gulley, J.L.; Schlom, J. Elevated serum soluble CD40 ligand in cancer patients may play an immunosuppressive role. Blood 2012, 120, 3030–3038. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef]
- Munn, D.H. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci. 2012, 4, 734–745. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Li, Y.; Gasmi, B.; Munn, D.H.; Allison, J.P.; Merghoub, T.; Wolchok, J.D. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015, 13, 412–424. [Google Scholar] [CrossRef]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef]
- Welte, T.; Kim, I.S.; Tian, L.; Gao, X.; Wang, H.; Li, J.; Holdman, X.B.; Herschkowitz, J.I.; Pond, A.; Xie, G.; et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat. Cell Biol. 2016, 18, 632–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Gu, Y.; Xue, Y.; Yuan, M.; Cao, X.; Liu, Q. CXCR2(+) MDSCs promote breast cancer progression by inducing EMT and activated T cell exhaustion. Oncotarget 2017, 8, 114554–114567. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef]
- Fessenden, T.B.; Duong, E.; Spranger, S. A team effort: Natural killer cells on the first leg of the tumor immunity relay race. J. Immunother. Cancer 2018, 6, 67. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Huergo-Zapico, L.; Parodi, M.; Cantoni, C.; Lavarello, C.; Fernandez-Martinez, J.L.; Petretto, A.; DeAndres-Galiana, E.J.; Balsamo, M.; Lopez-Soto, A.; Pietra, G.; et al. NK-cell Editing Mediates Epithelial-to-Mesenchymal Transition via Phenotypic and Proteomic Changes in Melanoma Cell Lines. Cancer Res. 2018, 78, 3913–3925. [Google Scholar] [CrossRef]
- Levi, I.; Amsalem, H.; Nissan, A.; Darash-Yahana, M.; Peretz, T.; Mandelboim, O.; Rachmilewitz, J. Characterization of tumor infiltrating natural killer cell subset. Oncotarget 2015, 6, 13835–13843. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Ferlazzo, G.; Albini, A.; Noonan, D.M. A think tank of TINK/TANKs: Tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J. Natl. Cancer Inst. 2014, 106, dju200. [Google Scholar] [CrossRef]
- Park, I.H.; Yang, H.N.; Lee, K.J.; Kim, T.S.; Lee, E.S.; Jung, S.Y.; Kwon, Y.; Kong, S.Y. Tumor-derived IL-18 induces PD-1 expression on immunosuppressive NK cells in triple-negative breast cancer. Oncotarget 2017, 8, 32722–32730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrien-Elliott, M.M.; Romee, R.; Fehniger, T.A. Improving natural killer cell cancer immunotherapy. Curr. Opin. Organ Transplant. 2015, 20, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef]
- Johansson, J.; Tabor, V.; Wikell, A.; Jalkanen, S.; Fuxe, J. TGF-beta1-Induced Epithelial-Mesenchymal Transition Promotes Monocyte/Macrophage Properties in Breast Cancer Cells. Front. Oncol. 2015, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Faraji, F.; Pang, Y.; Walker, R.C.; Nieves Borges, R.; Yang, L.; Hunter, K.W. Cadm1 is a metastasis susceptibility gene that suppresses metastasis by modifying tumor interaction with the cell-mediated immunity. PLoS Genet. 2012, 8, e1002926. [Google Scholar] [CrossRef] [PubMed]
- Chockley, P.J.; Chen, J.; Chen, G.; Beer, D.G.; Standiford, T.J.; Keshamouni, V.G. Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer. J. Clin. Investig. 2018, 128, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Halle, S.; Halle, O.; Forster, R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo. Trends Immunol. 2017, 38, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluhar, G.E.; Pennell, C.A.; Olin, M.R. CD8(+) T Cell-Independent Immune-Mediated Mechanisms of Anti-Tumor Activity. Crit. Rev. Immunol. 2015, 35, 153–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Nishikawa, H. Roles of regulatory T cells in cancer immunity. Int. Immunol. 2016, 28, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Xiong, S.; Pan, X.; Xu, L.; Yang, Z.; Guo, R.; Gu, Y.; Li, R.; Wang, Q.; Xiao, F.; Du, L.; et al. Regulatory T Cells Promote beta-Catenin—Mediated Epithelium-to-Mesenchyme Transition During Radiation-Induced Pulmonary Fibrosis. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 425–435. [Google Scholar] [CrossRef]
- Sommers, C.L.; Heckford, S.E.; Skerker, J.M.; Worland, P.; Torri, J.A.; Thompson, E.W.; Byers, S.W.; Gelmann, E.P. Loss of epithelial markers and acquisition of vimentin expression in adriamycin- and vinblastine-resistant human breast cancer cell lines. Cancer Res. 1992, 52, 5190–5197. [Google Scholar]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Muller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef]
- Chockley, P.J.; Keshamouni, V.G. Immunological Consequences of Epithelial-Mesenchymal Transition in Tumor Progression. J. Immunol. 2016, 197, 691–698. [Google Scholar] [CrossRef]
- Salvagno, C.; Ciampricotti, M.; Tuit, S.; Hau, C.S.; van Weverwijk, A.; Coffelt, S.B.; Kersten, K.; Vrijland, K.; Kos, K.; Ulas, T.; et al. Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat. Cell Biol. 2019, 21, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Ladoire, S.; Arnould, L.; Apetoh, L.; Coudert, B.; Martin, F.; Chauffert, B.; Fumoleau, P.; Ghiringhelli, F. Pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of tumor-infiltrating foxp3+ regulatory T cells. Clin. Cancer Res. 2008, 14, 2413–2420. [Google Scholar] [CrossRef]
- Antoon, J.W.; White, M.D.; Burow, M.E.; Beckman, B.S. Dual inhibition of sphingosine kinase isoforms ablates TNF-induced drug resistance. Oncol. Rep. 2012, 27, 1779–1786. [Google Scholar] [PubMed] [Green Version]
- Sukocheva, O.A. Expansion of Sphingosine Kinase and Sphingosine-1-Phosphate Receptor Function in Normal and Cancer Cells: From Membrane Restructuring to Mediation of Estrogen Signaling and Stem Cell Programming. Int. J. Mol. Sci. 2018, 19, 420. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage. metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Ouzounova, M.; Lee, E.; Piranlioglu, R.; El Andaloussi, A.; Kolhe, R.; Demirci, M.F.; Marasco, D.; Asm, I.; Chadli, A.; Hassan, K.A.; et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017, 8, 14979. [Google Scholar] [CrossRef] [Green Version]
- Arce Vargas, F.; Furness, A.J.S.; Solomon, I.; Joshi, K.; Mekkaoui, L.; Lesko, M.H.; Miranda Rota, E.; Dahan, R.; Georgiou, A.; Sledzinska, A.; et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 2017, 46, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Ohm, J.E.; Gabrilovich, D.I.; Sempowski, G.D.; Kisseleva, E.; Parman, K.S.; Nadaf, S.; Carbone, D.P. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 2003, 101, 4878–4886. [Google Scholar] [CrossRef] [Green Version]
- Incio, J.; Ligibel, J.A.; McManus, D.T.; Suboj, P.; Jung, K.; Kawaguchi, K.; Pinter, M.; Babykutty, S.; Chin, S.M.; Vardam, T.D.; et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Luque-Cabal, M.; García-Teijido, P.; Fernández-Pérez, Y.; Sánchez-Lorenzo, L.; Palacio-Vázquez, I. Mechanisms Behind the Resistance to Trastuzumab in HER2-Amplified Breast Cancer and Strategies to Overcome It. Clin. Med. Insights Oncol. 2016, 10, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Barrett, M.T.; Lenkiewicz, E.; Malasi, S.; Basu, A.; Yearley, J.H.; Annamalai, L.; McCullough, A.E.; Kosiorek, H.E.; Narang, P.; Wilson Sayres, M.A.; et al. The association of genomic lesions and PD-1/PD-L1 expression in resected triple-negative breast cancers. Breast Cancer Res. 2018, 20, 71. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef]
- Muenst, S.; Soysal, S.D.; Gao, F.; Obermann, E.C.; Oertli, D.; Gillanders, W.E. The presence of programmed death 1 (PD-1)-positive tumor-infiltrating lymphocytes is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2013, 139, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef] [PubMed]
- Nicolazzo, C.; Raimondi, C.; Mancini, M.; Caponnetto, S.; Gradilone, A.; Gandini, O.; Mastromartino, M.; Del Bene, G.; Prete, A.; Longo, F.; et al. Monitoring PD-L1 positive circulating tumor cells in non-small cell lung cancer patients treated with the PD-1 inhibitor Nivolumab. Sci. Rep. 2016, 6, 31726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, C.; Carpino, G.; Nicolazzo, C.; Gradilone, A.; Gianni, W.; Gelibter, A.; Gaudio, E.; Cortesi, E.; Gazzaniga, P. PD-L1 and epithelial-mesenchymal transition in circulating tumor cells from non-small cell lung cancer patients: A molecular shield to evade immune system? Oncoimmunology 2017, 6, e1315488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Kitamura, T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 38. [Google Scholar] [CrossRef]
- Janakiram, M.; Abadi, Y.M.; Sparano, J.A.; Zang, X. T cell coinhibition and immunotherapy in human breast cancer. Discov. Med. 2012, 14, 229–236. [Google Scholar] [PubMed]


{kind=link}
| Breast Cancer Subtypes | Genes Involved | Signaling Pathways Involved | References |
|---|---|---|---|
| Luminal A and B | SLUG, BCL2, BM1, TGF-β, TWIST, ZEB1/2, ETS1, VEGFR | ERα signaling, TGF-β signaling, EGFR-, IGFR-, and MAPK-dependent, MEK–ERK | [36,37,38,39,40,41,42,43,44,45,46] |
| HER2-positive | TGF-β, TWIST1, PTK-6 | TGF-β signaling, AKT signaling, HER2/EGFR signaling | [47,48,49,50,52,53,54] |
| TNBC (Basal and Claudin-Low) | TGF-β, GLI1, SNAIL, SLUG, TWIST1, ZEB1, ΔNP63, GSK3β | PI3K/AKT, Notch signaling, Hedgehog signaling, Wnt signaling | [59,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Chakrabarti, R. Consequences of EMT-Driven Changes in the Immune Microenvironment of Breast Cancer and Therapeutic Response of Cancer Cells. J. Clin. Med. 2019, 8, 642. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050642
Singh S, Chakrabarti R. Consequences of EMT-Driven Changes in the Immune Microenvironment of Breast Cancer and Therapeutic Response of Cancer Cells. Journal of Clinical Medicine. 2019; 8(5):642. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050642
Chicago/Turabian StyleSingh, Snahlata, and Rumela Chakrabarti. 2019. "Consequences of EMT-Driven Changes in the Immune Microenvironment of Breast Cancer and Therapeutic Response of Cancer Cells" Journal of Clinical Medicine 8, no. 5: 642. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050642