Phosphoproteomic Profiling Identifies Aberrant Activation of Integrin Signaling in Aggressive Non-Type Bladder Carcinoma

Abstract
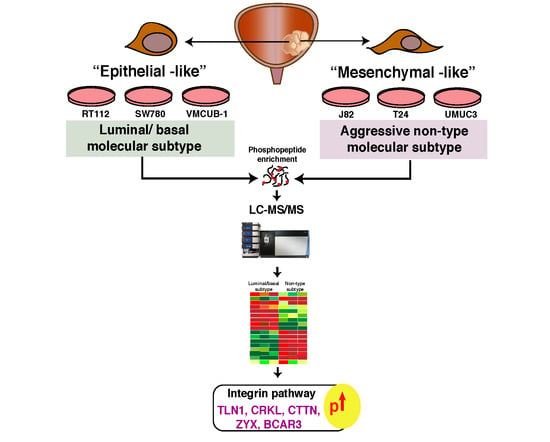
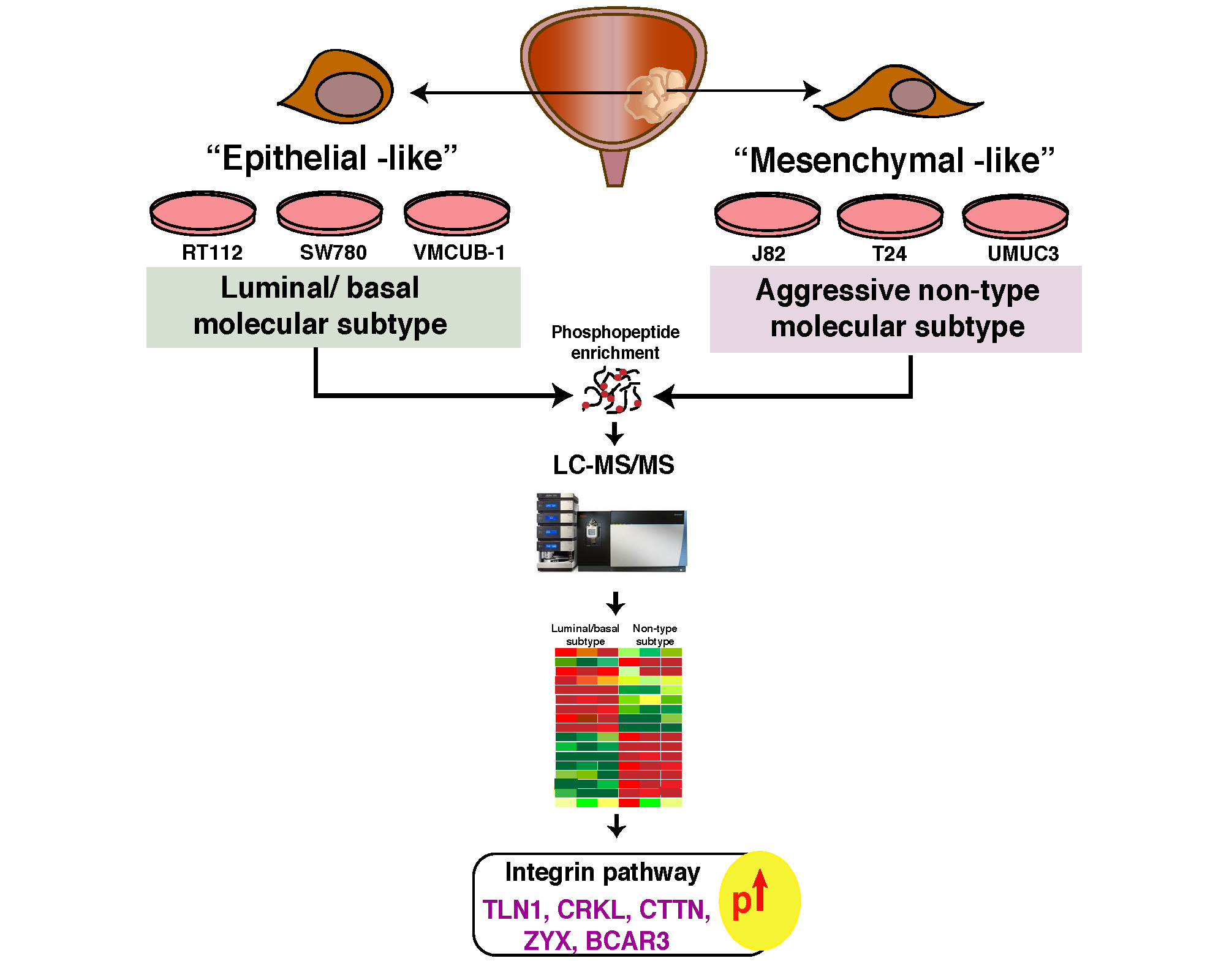
:
1. Introduction
2. Experimental Section
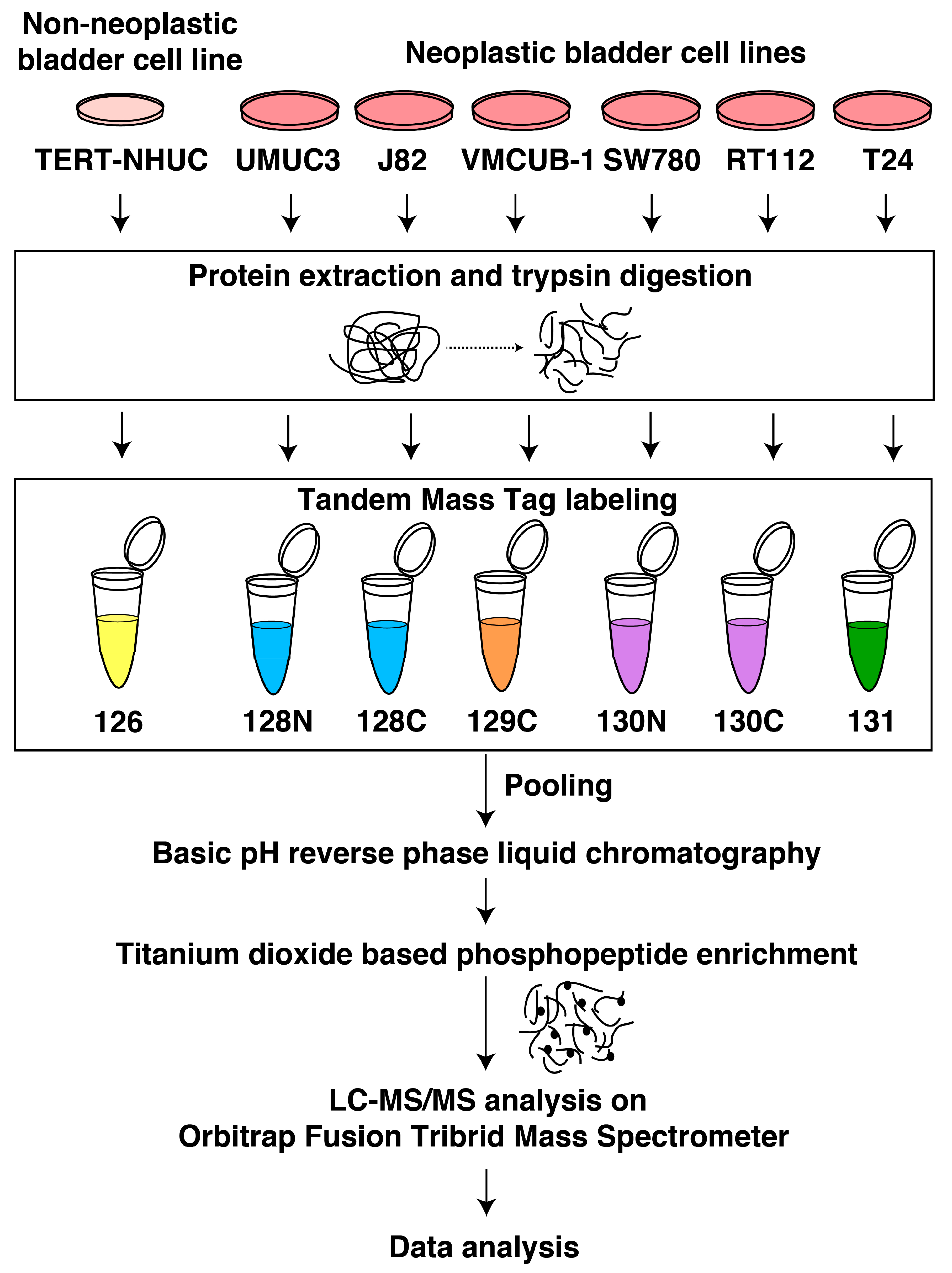
2.1. Cell Culture
2.2. Cell Lysis, Protein Extraction, and Digestion
2.3. TMT Labeling
2.4. Basic pH RPLC (bRPLC) and TiO2-Based Phosphopeptide Enrichment
2.5. LC-MS/MS Analysis
2.6. Data Analysis
2.7. Scratch Wound Assay
2.8. Invasion Assay
2.9. Kinome Map
2.10. Clustering the Molecular Subtypes
2.11. Ingenuity Pathway Analysis
2.12. Kinase-Substrate Enrichment Analysis
2.13. Motif Analysis
2.14. Immunohistochemistry
3. Results
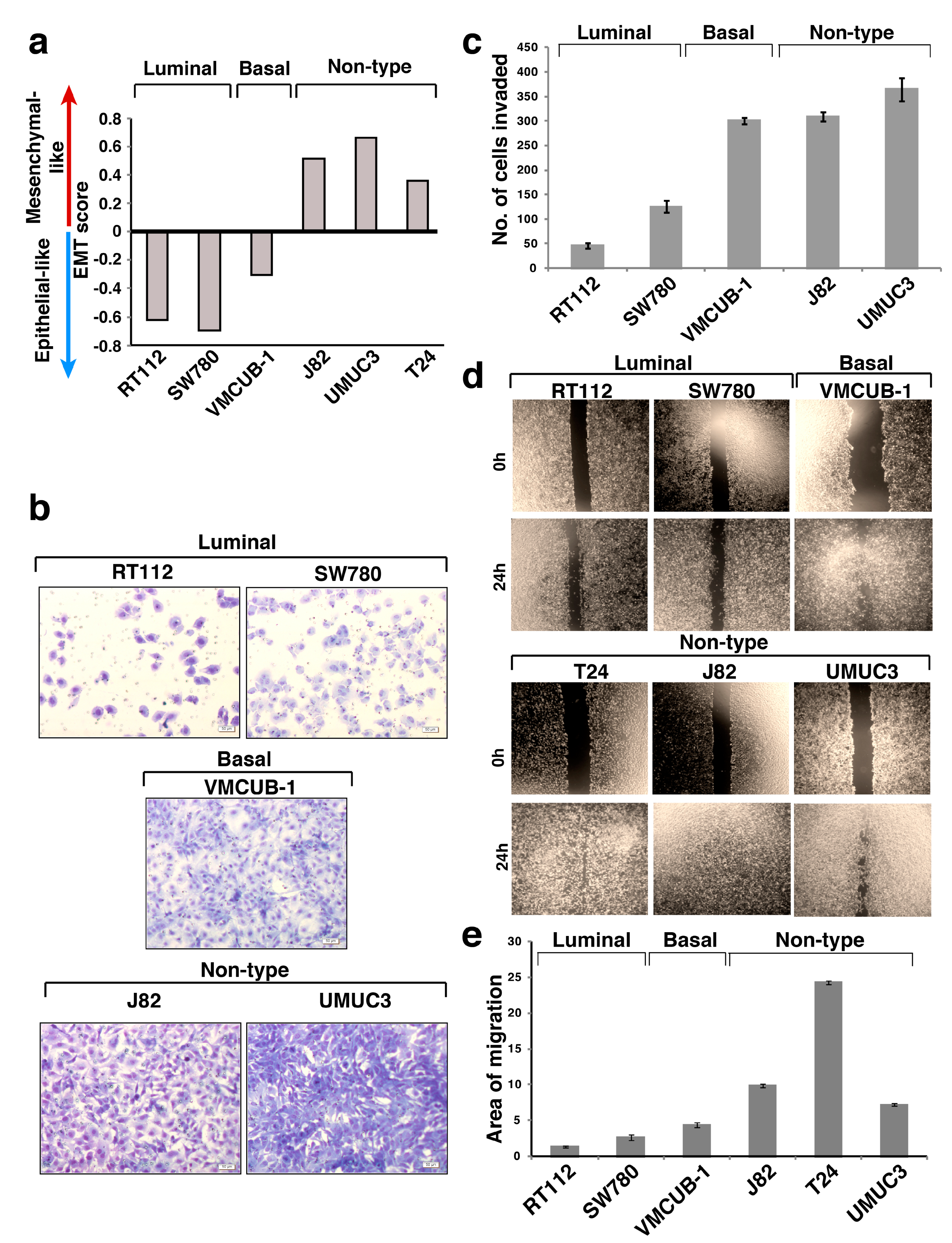
3.1. EMT Scores of the Molecular Subtypes of Bladder Carcinoma Cell Lines
3.2. Non-Type Bladder Carcinoma Has Increased Migration and Invasive Ability
3.3. Phosphoproteomic Analysis of Bladder Carcinoma Cell Lines
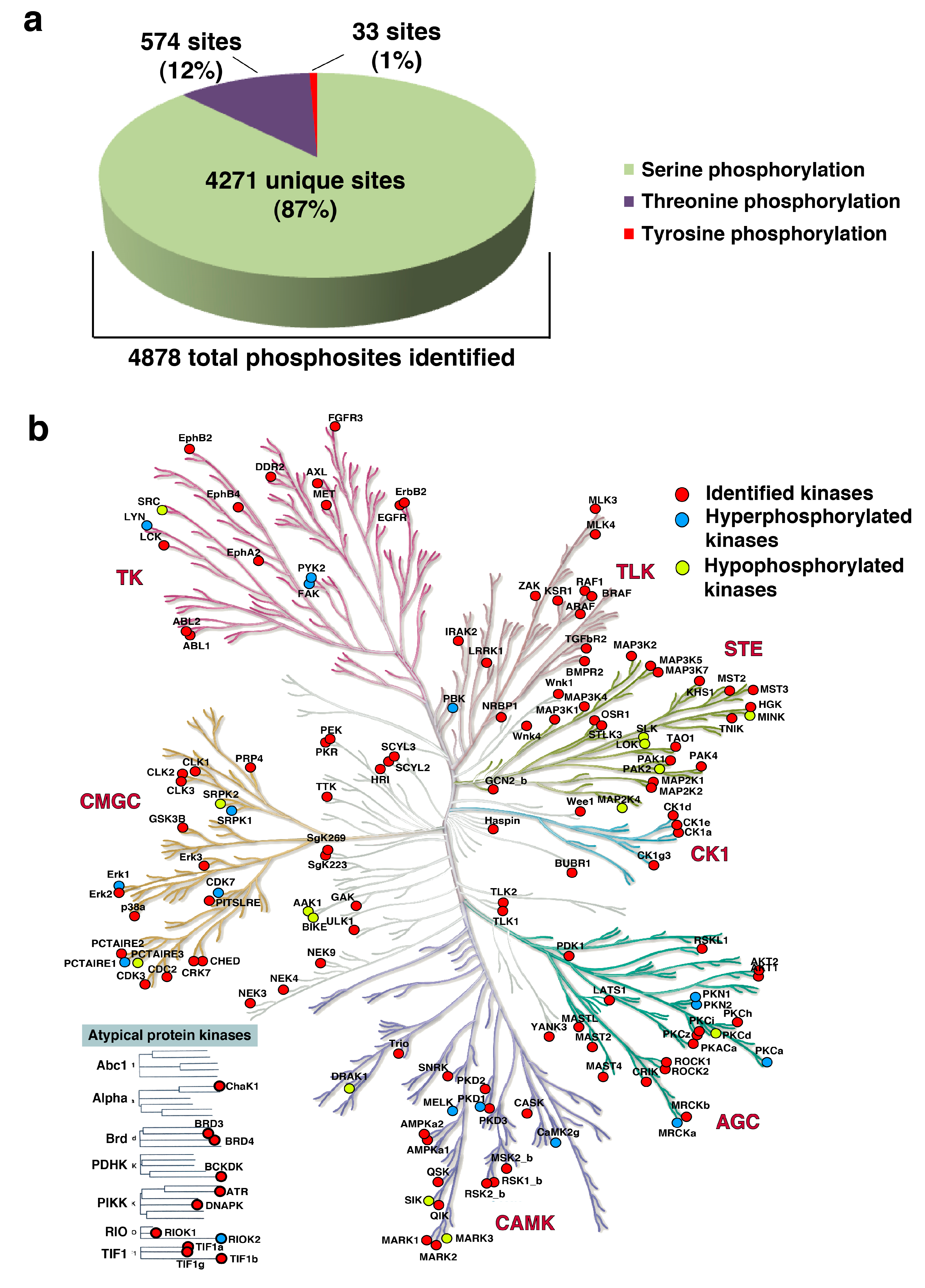
3.4. Kinases Enriched in the Phosphoproteomics Dataset
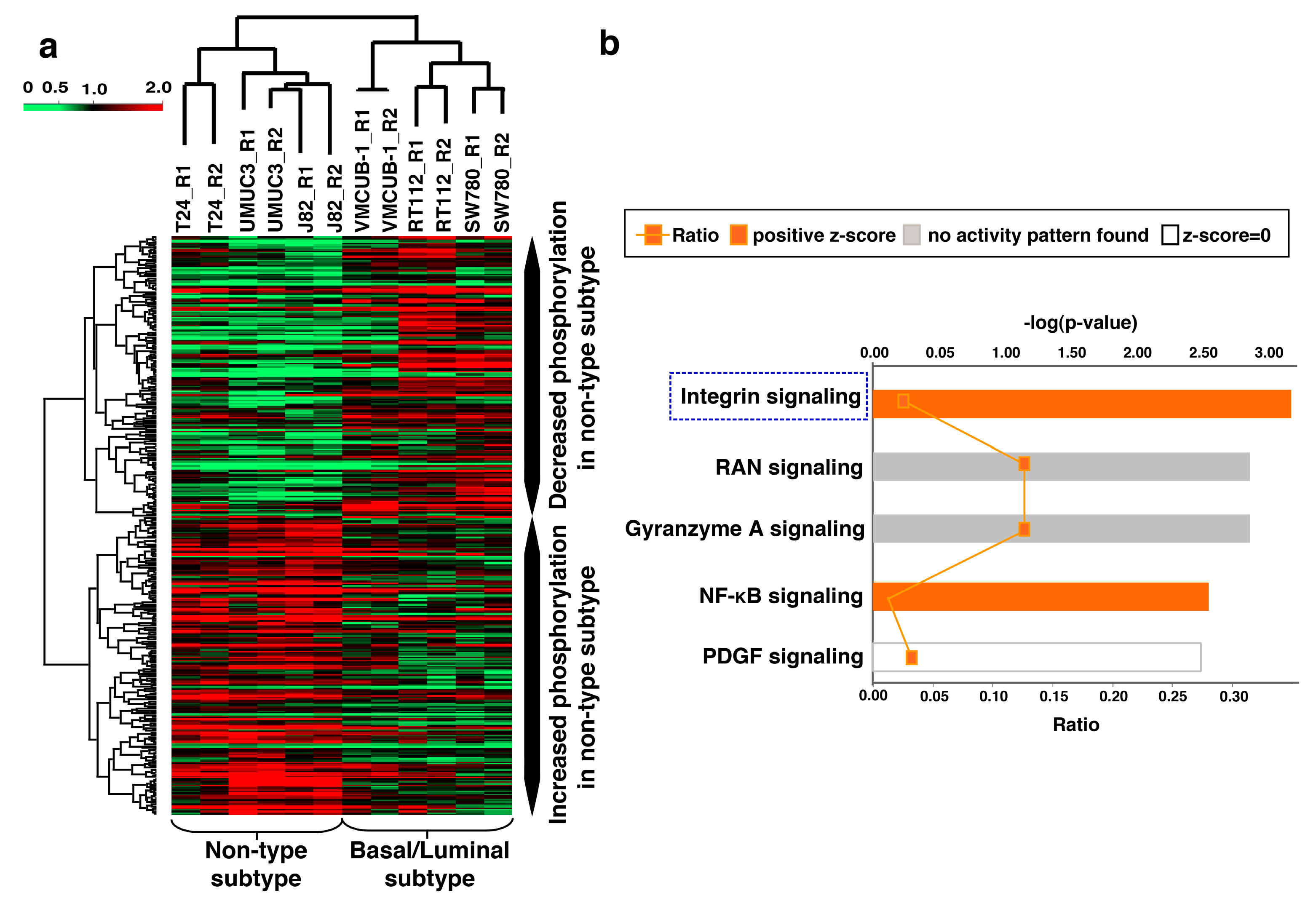
3.5. Unique Phosphorylation Signature Identified for the Non-Type Subtype of Bladder Carcinoma
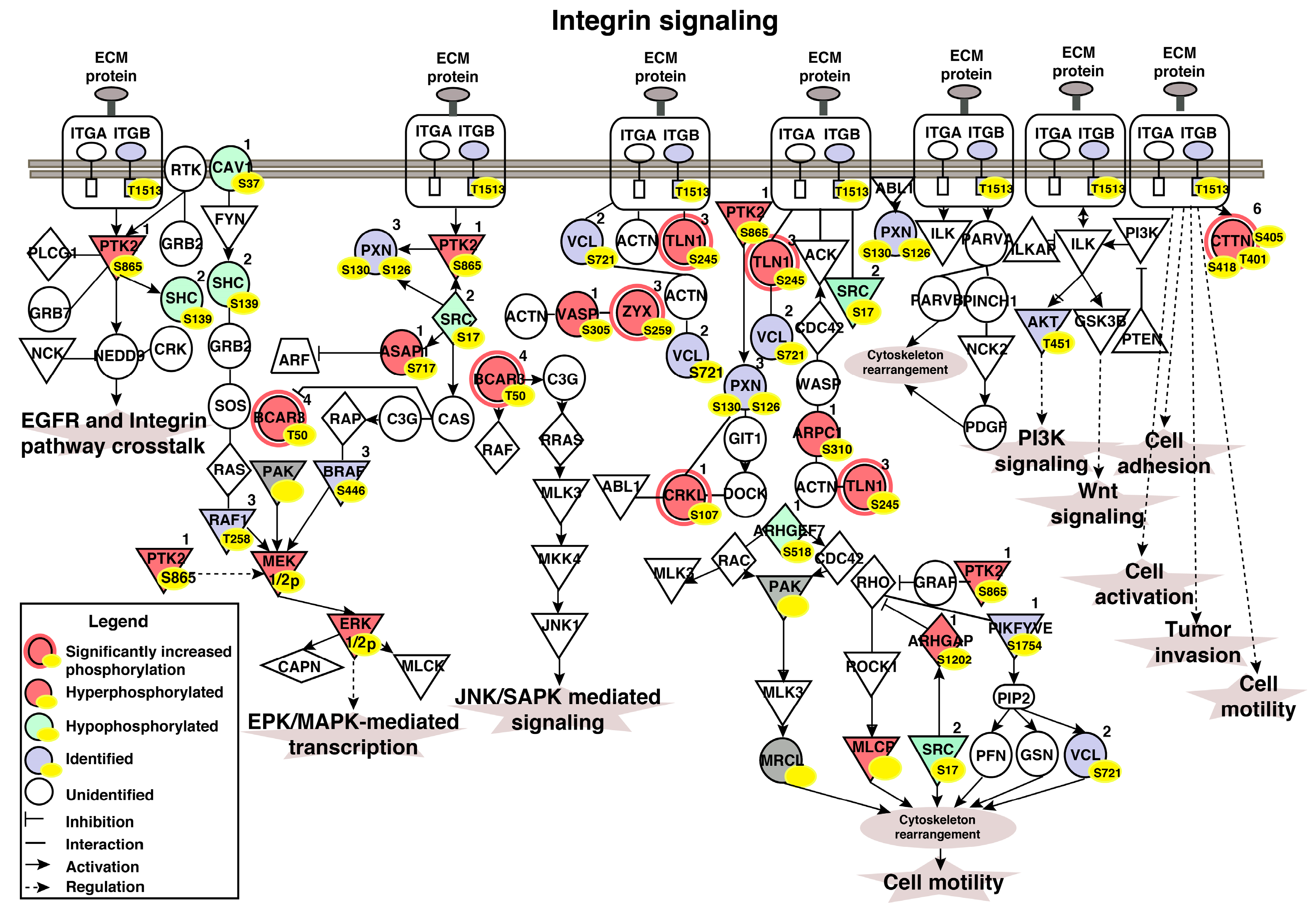
3.6. Ingenuity Pathway Analysis (IPA) Identifies Aberrant Activation of Pathways in the Molecular Subtypes of Bladder Carcinoma
3.7. Regulatory Interaction Network Enriched in the Non-Type Subtype
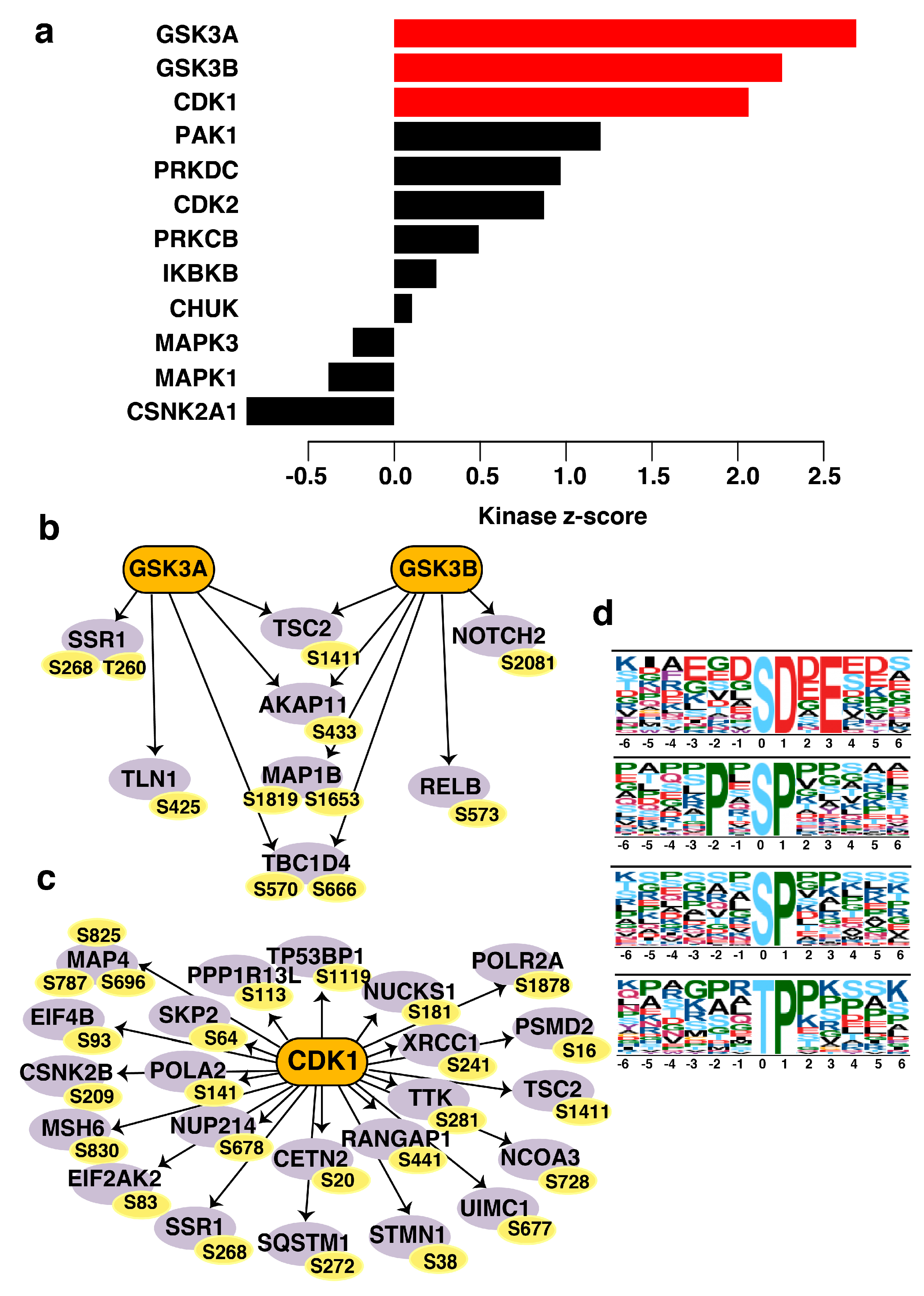
3.8. CDK1 (Cyclin-Dependent Kinase 1) and GSK3A/GSK3B (Glycogen Synthase Kinase 3A and 3B) Are the Predicted Activated Kinases in Non-Type Subtype of Bladder Carcinoma Cell Lines
3.9. Active Proline-Directed Motifs Identified in the Non-Type Molecular Subtype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burger, M.; Oosterlinck, W.; Konety, B.; Chang, S.; Gudjonsson, S.; Pruthi, R.; Soloway, M.; Solsona, E.; Sved, P.; Babjuk, M.; et al. ICUD-EAU International Consultation on Bladder Cancer 2012: Non-muscle-invasive urothelial carcinoma of the bladder. Eur. Urol. 2013, 63, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Schrier, B.P.; Hollander, M.P.; van Rhijn, B.W.; Kiemeney, L.A.; Witjes, J.A. Prognosis of muscle-invasive bladder cancer: Difference between primary and progressive tumours and implications for therapy. Eur. Urol. 2004, 45, 292–296. [Google Scholar] [CrossRef]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef] [Green Version]
- Damrauer, J.S.; Hoadley, K.A.; Chism, D.D.; Fan, C.; Tiganelli, C.J.; Wobker, S.E.; Yeh, J.J.; Milowsky, M.I.; Iyer, G.; Parker, J.S.; et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc. Natl. Acad. Sci. USA 2014, 111, 3110–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.L.; et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, K.; Yamada, Y.; Okajima, E.; Kakizoe, T.; Sasaki, H.; Sugimura, T.; Terada, M. Frequent association of p53 gene mutation in invasive bladder cancer. Cancer Res. 1992, 52, 1393–1398. [Google Scholar] [PubMed]
- Van Rhijn, B.W.; Lurkin, I.; Radvanyi, F.; Kirkels, W.J.; van der Kwast, T.H.; Zwarthoff, E.C. The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Res. 2001, 61, 1265–1268. [Google Scholar] [PubMed]
- Orlow, I.; Lacombe, L.; Hannon, G.J.; Serrano, M.; Pellicer, I.; Dalbagni, G.; Reuter, V.E.; Zhang, Z.F.; Beach, D.; Cordon-Cardo, C. Deletion of the p16 and p15 genes in human bladder tumors. J. Natl. Cancer Inst. 1995, 87, 1524–1529. [Google Scholar] [CrossRef]
- Sjodahl, G.; Lauss, M.; Lovgren, K.; Chebil, G.; Gudjonsson, S.; Veerla, S.; Patschan, O.; Aine, M.; Ferno, M.; Ringner, M.; et al. A molecular taxonomy for urothelial carcinoma. Clin. Cancer Res. 2012, 18, 3377–3386. [Google Scholar] [CrossRef] [PubMed]
- Aine, M.; Eriksson, P.; Liedberg, F.; Sjodahl, G.; Hoglund, M. Biological determinants of bladder cancer gene expression subtypes. Sci. Rep. 2015, 5, 10957. [Google Scholar] [CrossRef] [Green Version]
- Hedegaard, J.; Lamy, P.; Nordentoft, I.; Algaba, F.; Hoyer, S.; Ulhoi, B.P.; Vang, S.; Reinert, T.; Hermann, G.G.; Mogensen, K.; et al. Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell 2016, 30, 27–42. [Google Scholar] [CrossRef]
- Svatek, R.S.; Shariat, S.F.; Novara, G.; Skinner, E.C.; Fradet, Y.; Bastian, P.J.; Kamat, A.M.; Kassouf, W.; Karakiewicz, P.I.; Fritsche, H.M.; et al. Discrepancy between clinical and pathological stage: External validation of the impact on prognosis in an international radical cystectomy cohort. BJU Int. 2011, 107, 898–904. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Warrick, J.I.; Walter, V.; Yamashita, H.; Chung, E.; Shuman, L.; Amponsa, V.O.; Zheng, Z.; Chan, W.; Whitcomb, T.L.; Yue, F.; et al. FOXA1, GATA3 and PPAR Cooperate to Drive Luminal Subtype in Bladder Cancer: A Molecular Analysis of Established Human Cell Lines. Sci. Rep. 2016, 6, 38531. [Google Scholar] [CrossRef]
- Chapman, E.J.; Hurst, C.D.; Pitt, E.; Chambers, P.; Aveyard, J.S.; Knowles, M.A. Expression of hTERT immortalises normal human urothelial cells without inactivation of the p16/Rb pathway. Oncogene 2006, 25, 5037–5045. [Google Scholar] [CrossRef] [Green Version]
- Selvan, L.D.; Renuse, S.; Kaviyil, J.E.; Sharma, J.; Pinto, S.M.; Yelamanchi, S.D.; Puttamallesh, V.N.; Ravikumar, R.; Pandey, A.; Prasad, T.S.; et al. Phosphoproteome of Cryptococcus neoformans. J. Proteomics 2014, 97, 287–295. [Google Scholar] [CrossRef]
- Pinto, S.M.; Nirujogi, R.S.; Rojas, P.L.; Patil, A.H.; Manda, S.S.; Subbannayya, Y.; Roa, J.C.; Chatterjee, A.; Prasad, T.S.; Pandey, A. Quantitative phosphoproteomic analysis of IL-33-mediated signaling. Proteomics 2015, 15, 532–544. [Google Scholar] [CrossRef]
- Verma, R.; Pinto, S.M.; Patil, A.H.; Advani, J.; Subba, P.; Kumar, M.; Sharma, J.; Dey, G.; Ravikumar, R.; Buggi, S.; et al. Quantitative Proteomic and Phosphoproteomic Analysis of H37Ra and H37Rv Strains of Mycobacterium tuberculosis. J. Proteome Res. 2017, 16, 1632–1645. [Google Scholar] [CrossRef]
- Subbannayya, Y.; Syed, N.; Barbhuiya, M.A.; Raja, R.; Marimuthu, A.; Sahasrabuddhe, N.; Pinto, S.M.; Manda, S.S.; Renuse, S.; Manju, H.C.; et al. Calcium calmodulin dependent kinase kinase 2-a novel therapeutic target for gastric adenocarcinoma. Cancer Biol. Ther. 2015, 16, 336–345. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Wiredja, D.D.; Koyuturk, M.; Chance, M.R. The KSEA App: A web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics 2017. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Invest. 2013, 123, 517–525. [Google Scholar] [CrossRef]
- Gatza, M.L.; Lucas, J.E.; Barry, W.T.; Kim, J.W.; Wang, Q.; Crawford, M.D.; Datto, M.B.; Kelley, M.; Mathey-Prevot, B.; Potti, A.; et al. A pathway-based classification of human breast cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 6994–6999. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Ithychanda, S.; Qin, J.; Plow, E.F. Mechanisms of talin-dependent integrin signaling and crosstalk. Biochim. Biophys. Acta 2014, 1838, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Vuori, K.; Ruoslahti, E. Tyrosine phosphorylation of p130Cas and cortactin accompanies integrin-mediated cell adhesion to extracellular matrix. J. Biol. Chem. 1995, 270, 22259–22262. [Google Scholar] [CrossRef]
- Li, L.; Guris, D.L.; Okura, M.; Imamoto, A. Translocation of CrkL to focal adhesions mediates integrin-induced migration downstream of Src family kinases. Mol. Cell Biol. 2003, 23, 2883–2892. [Google Scholar] [CrossRef]
- Xu, C.; Yang, Y.; Yang, J.; Chen, X.; Wang, G. Analysis of the role of the integrin signaling pathway in hepatocytes during rat liver regeneration. Cell Mol. Biol. Lett. 2012, 17, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Green, Y.S.; Kwon, S.; Christian, J.L. Expression pattern of bcar3, a downstream target of Gata2, and its binding partner, bcar1, during Xenopus development. Gene Expr. Patterns 2016, 20, 55–62. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2019, 194, 161–184. [Google Scholar] [CrossRef]
- Yadavalli, S.; Jayaram, S.; Manda, S.S.; Madugundu, A.K.; Nayakanti, D.S.; Tan, T.Z.; Bhat, R.; Rangarajan, A.; Chatterjee, A.; Gowda, H.; et al. Data-Driven Discovery of Extravasation Pathway in Circulating Tumor Cells. Sci. Rep. 2017, 7, 43710. [Google Scholar] [CrossRef] [Green Version]
- Taherian, A.; Li, X.; Liu, Y.; Haas, T.A. Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer 2011, 11, 293. [Google Scholar] [CrossRef]
- Boudjadi, S.; Carrier, J.C.; Beaulieu, J.F. Integrin alpha1 subunit is up-regulated in colorectal cancer. Biomark. Res. 2013, 1, 16. [Google Scholar] [CrossRef]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.D.; Cheresh, D.A. Role of integrins in cell invasion and migration. Nat. Rev. Cancer 2002, 2, 91–100. [Google Scholar] [CrossRef]
- Radeva, G.; Petrocelli, T.; Behrend, E.; Leung-Hagesteijn, C.; Filmus, J.; Slingerland, J.; Dedhar, S. Overexpression of the integrin-linked kinase promotes anchorage-independent cell cycle progression. J. Biol. Chem. 1997, 272, 13937–13944. [Google Scholar] [CrossRef]
- Le Gall, M.; Grall, D.; Chambard, J.C.; Pouyssegur, J.; Van Obberghen-Schilling, E. An anchorage-dependent signal distinct from p42/44 MAP kinase activation is required for cell cycle progression. Oncogene 1998, 17, 1271–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.K.; Tien, P.C.; Cheng, C.J.; Song, J.H.; Huang, C.; Lin, S.H.; Gallick, G.E. Talin1 phosphorylation activates beta1 integrins: A novel mechanism to promote prostate cancer bone metastasis. Oncogene 2015, 34, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Strock, C.J.; Park, J.I.; Nakakura, E.K.; Bova, G.S.; Isaacs, J.T.; Ball, D.W.; Nelkin, B.D. Cyclin-dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer Res. 2006, 66, 7509–7515. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, G.; Cai, Y.; Monkley, S.J.; Critchley, D.R.; Sheetz, M.P. Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat. Cell Biol. 2008, 10, 1062–1068. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Rajfur, Z.; Yousefi, N.; Chen, Z.; Jacobson, K.; Ginsberg, M.H. Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nat. Cell Biol. 2009, 11, 624–630. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.T.; Hua, C.H.; Tsai, M.H.; Wan, L.; Lin, Y.J.; Chen, C.M.; Chiu, I.W.; Chan, C.; Tsai, F.J.; Jinn-Chyuan Sheu, J. Talin-1 overexpression defines high risk for aggressive oral squamous cell carcinoma and promotes cancer metastasis. J. Pathol. 2011, 224, 367–376. [Google Scholar] [CrossRef]
- He, Y.; Ren, Y.; Wu, B.; Decourt, B.; Lee, A.C.; Taylor, A.; Suter, D.M. Src and cortactin promote lamellipodia protrusion and filopodia formation and stability in growth cones. Mol. Biol. Cell 2015, 26, 3229–3244. [Google Scholar] [CrossRef] [Green Version]
- Kruchten, A.E.; Krueger, E.W.; Wang, Y.; McNiven, M.A. Distinct phospho-forms of cortactin differentially regulate actin polymerization and focal adhesions. Am. J. Physiol. Cell Physiol. 2008, 295, C1113–C1122. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Quiles, N.; Ho, H.Y.; Kirschner, M.W.; Ramesh, N.; Geha, R.S. Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP. Mol. Cell Biol. 2004, 24, 5269–5280. [Google Scholar] [CrossRef]
- Kelley, L.C.; Hayes, K.E.; Ammer, A.G.; Martin, K.H.; Weed, S.A. Cortactin phosphorylated by ERK1/2 localizes to sites of dynamic actin regulation and is required for carcinoma lamellipodia persistence. PLoS ONE 2010, 5, e13847. [Google Scholar] [CrossRef]
- Tokui, N.; Yoneyama, M.S.; Hatakeyama, S.; Yamamoto, H.; Koie, T.; Saitoh, H.; Yamaya, K.; Funyu, T.; Nakamura, T.; Ohyama, C.; et al. Extravasation during bladder cancer metastasis requires cortactinmediated invadopodia formation. Mol. Med. Rep. 2014, 9, 1142–1146. [Google Scholar] [CrossRef]
- Han, B.; Luan, L.; Xu, Z.; Wu, B. Clinical significance and biological roles of CRKL in human bladder carcinoma. Tumour. Biol. 2014, 35, 4101–4106. [Google Scholar] [CrossRef]
- Mise, N.; Savai, R.; Yu, H.; Schwarz, J.; Kaminski, N.; Eickelberg, O. Zyxin is a transforming growth factor-beta (TGF-beta)/Smad3 target gene that regulates lung cancer cell motility via integrin alpha5beta1. J. Biol. Chem. 2012, 287, 31393–31405. [Google Scholar] [CrossRef]
- Ma, B.; Cheng, H.; Gao, R.; Mu, C.; Chen, L.; Wu, S.; Chen, Q.; Zhu, Y. Zyxin-Siah2-Lats2 axis mediates cooperation between Hippo and TGF-beta signalling pathways. Nat. Commun. 2016, 7, 11123. [Google Scholar] [CrossRef] [PubMed]
- Riggins, R.B.; Quilliam, L.A.; Bouton, A.H. Synergistic promotion of c-Src activation and cell migration by Cas and AND-34/BCAR3. J. Biol. Chem. 2003, 278, 28264–28273. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Cheng, S.Y.; Chen, M.; Lim, C.J.; Pallen, C.J. Protein tyrosine phosphatase alpha phosphotyrosyl-789 binds BCAR3 to position Cas for activation at integrin-mediated focal adhesions. Mol. Cell Biol. 2012, 32, 3776–3789. [Google Scholar] [CrossRef]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef]
- Roca, H.; Hernandez, J.; Weidner, S.; McEachin, R.C.; Fuller, D.; Sud, S.; Schumann, T.; Wilkinson, J.E.; Zaslavsky, A.; Li, H.; et al. Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS ONE 2013, 8, e76773. [Google Scholar] [CrossRef]
- Feng, J.; Xu, G.; Liu, J.; Zhang, N.; Li, L.; Ji, J.; Zhang, J.; Zhang, L.; Wang, G.; Wang, X.; et al. Phosphorylation of LSD1 at Ser112 is crucial for its function in induction of EMT and metastasis in breast cancer. Breast Cancer Res. Treat. 2016, 159, 443–456. [Google Scholar] [CrossRef]
- Sun, L.; Fang, J. Epigenetic regulation of epithelial-mesenchymal transition. Cell Mol. Life Sci. 2016, 73, 4493–4515. [Google Scholar] [CrossRef] [PubMed]
- Warns, J.A.; Davie, J.R.; Dhasarathy, A. Connecting the dots: Chromatin and alternative splicing in EMT. Biochem. Cell Biol. 2016, 94, 12–25. [Google Scholar] [CrossRef]
- Ikuta, S.; Edamatsu, H.; Li, M.; Hu, L.; Kataoka, T. Crucial role of phospholipase C epsilon in skin inflammation induced by tumor-promoting phorbol ester. Cancer Res. 2008, 68, 64–72. [Google Scholar] [CrossRef]
- Chen, J.; Wang, W.; Zhang, T.; Ji, J.; Qian, Q.; Lu, L.; Fu, H.; Jin, W.; Cui, D. Differential expression of phospholipase C epsilon 1 is associated with chronic atrophic gastritis and gastric cancer. PLoS ONE 2012, 7, e47563. [Google Scholar] [CrossRef]
- Burbelo, P.; Wellstein, A.; Pestell, R.G. Altered Rho GTPase signaling pathways in breast cancer cells. Breast Cancer Res. Treat. 2004, 84, 43–48. [Google Scholar] [CrossRef]
- Kamai, T.; Tsujii, T.; Arai, K.; Takagi, K.; Asami, H.; Ito, Y.; Oshima, H. Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin. Cancer Res. 2003, 9, 2632–2641. [Google Scholar]
- Dadhania, V.; Zhang, M.; Zhang, L.; Bondaruk, J.; Majewski, T.; Siefker-Radtke, A.; Guo, C.C.; Dinney, C.; Cogdell, D.E.; Zhang, S.; et al. Meta-Analysis of the Luminal and Basal Subtypes of Bladder Cancer and the Identification of Signature Immunohistochemical Markers for Clinical Use. EBioMedicine 2016, 12, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-kappaB addiction and its role in cancer: ‘One size does not fit all’. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef]
- Ahmed, K.M.; Zhang, H.; Park, C.C. NF-kappaB regulates radioresistance mediated by beta1-integrin in three-dimensional culture of breast cancer cells. Cancer Res. 2013, 73, 3737–3748. [Google Scholar] [CrossRef]
- Leis, H.; Segrelles, C.; Ruiz, S.; Santos, M.; Paramio, J.M. Expression, localization, and activity of glycogen synthase kinase 3beta during mouse skin tumorigenesis. Mol. Carcinog. 2002, 35, 180–185. [Google Scholar] [CrossRef]
- Caccavari, F.; Valdembri, D.; Sandri, C.; Bussolino, F.; Serini, G. Integrin signaling and lung cancer. Cell Adh. Migr. 2010, 4, 124–129. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [Green Version]
- Panka, D.J.; Cho, D.C.; Atkins, M.B.; Mier, J.W. GSK-3beta inhibition enhances sorafenib-induced apoptosis in melanoma cell lines. J. Biol. Chem. 2008, 283, 726–732. [Google Scholar] [CrossRef]
- Mirlashari, M.R.; Randen, I.; Kjeldsen-Kragh, J. Glycogen synthase kinase-3 (GSK-3) inhibition induces apoptosis in leukemic cells through mitochondria-dependent pathway. Leuk. Res. 2012, 36, 499–508. [Google Scholar] [CrossRef]
- Cao, Q.; Lu, X.; Feng, Y.J. Glycogen synthase kinase-3beta positively regulates the proliferation of human ovarian cancer cells. Cell. Res. 2006, 16, 671–677. [Google Scholar] [CrossRef]
- Kang, J.; Sergio, C.M.; Sutherland, R.L.; Musgrove, E.A. Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells. BMC Cancer 2014, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Zeestraten, E.C.; Maak, M.; Shibayama, M.; Schuster, T.; Nitsche, U.; Matsushima, T.; Nakayama, S.; Gohda, K.; Friess, H.; van de Velde, C.J.; et al. Specific activity of cyclin-dependent kinase I is a new potential predictor of tumour recurrence in stage II colon cancer. Br. J. Cancer 2012, 106, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, F.H.; Chen, Q.E.; Wang, Y.Y.; Wang, Y.L.; He, J.C.; Zhou, J. The clinical significance of CDK1 expression in oral squamous cell carcinoma. Med. Oral Patol. Oral Cir. Bucal 2015, 20, e7–e12. [Google Scholar] [CrossRef] [PubMed]
- De Azevedo, W.F., Jr.; Mueller-Dieckmann, H.J.; Schulze-Gahmen, U.; Worland, P.J.; Sausville, E.; Kim, S.H. Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 2735–2740. [Google Scholar] [CrossRef] [PubMed]
- Christian, B.A.; Grever, M.R.; Byrd, J.C.; Lin, T.S. Flavopiridol in the treatment of chronic lymphocytic leukemia. Curr. Opin. Oncol. 2007, 19, 573–578. [Google Scholar] [CrossRef]
- Ghia, P.; Scarfo, L.; Perez, S.; Pathiraja, K.; Derosier, M.; Small, K.; McCrary Sisk, C.; Patton, N. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017, 129, 1876–1878. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.F.; Wang, R.; Liang, Q.; Liang, J.; Li, W.; Jung, S.Y.; Qin, J.; Lin, S.H.; Kuang, J. Dissecting the M phase-specific phosphorylation of serine-proline or threonine-proline motifs. Mol. Biol. Cell 2010, 21, 1470–1481. [Google Scholar] [CrossRef]
- Villen, J.; Beausoleil, S.A.; Gerber, S.A.; Gygi, S.P. Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. USA 2007, 104, 1488–1493. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Source a | Molecular Subtype b | Derived from Male/Female | Grade |
|---|---|---|---|---|
| SW780 | UCC | Luminal | Female | Grade 1 |
| RT112 | UCC | Luminal | Female | Grade 2 |
| T24 | EC | Non-type | Female | Grade 3 |
| J82 | EC | Non-type | Male | Grade 3 |
| UMUC3 | UCC | Non-type | Male | - |
| VMCUB-1 | EC | Basal | Male | Grade 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deb, B.; Puttamallesh, V.N.; Gondkar, K.; Thiery, J.P.; Gowda, H.; Kumar, P. Phosphoproteomic Profiling Identifies Aberrant Activation of Integrin Signaling in Aggressive Non-Type Bladder Carcinoma. J. Clin. Med. 2019, 8, 703. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050703
Deb B, Puttamallesh VN, Gondkar K, Thiery JP, Gowda H, Kumar P. Phosphoproteomic Profiling Identifies Aberrant Activation of Integrin Signaling in Aggressive Non-Type Bladder Carcinoma. Journal of Clinical Medicine. 2019; 8(5):703. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050703
Chicago/Turabian StyleDeb, Barnali, Vinuth N. Puttamallesh, Kirti Gondkar, Jean P. Thiery, Harsha Gowda, and Prashant Kumar. 2019. "Phosphoproteomic Profiling Identifies Aberrant Activation of Integrin Signaling in Aggressive Non-Type Bladder Carcinoma" Journal of Clinical Medicine 8, no. 5: 703. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050703