Computational Simulations Identify Pyrrolidine-2,3-Dione Derivatives as Novel Inhibitors of Cdk5/p25 Complex to Attenuate Alzheimer’s Pathology



 ,
,  ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Data Set Preparation and Pharmacophore Model Generation
2.2. Validation of Pharmacophore Model
2.3. Drug-Like Database Designing and Virtual Screening
2.4. Molecular Docking Simulation
2.5. Molecular Dynamics (MD) Simulation
2.6. Binding Free Energy Calculations
3. Results and Discussion
3.1. Ligand-Based Pharmacophore Generation
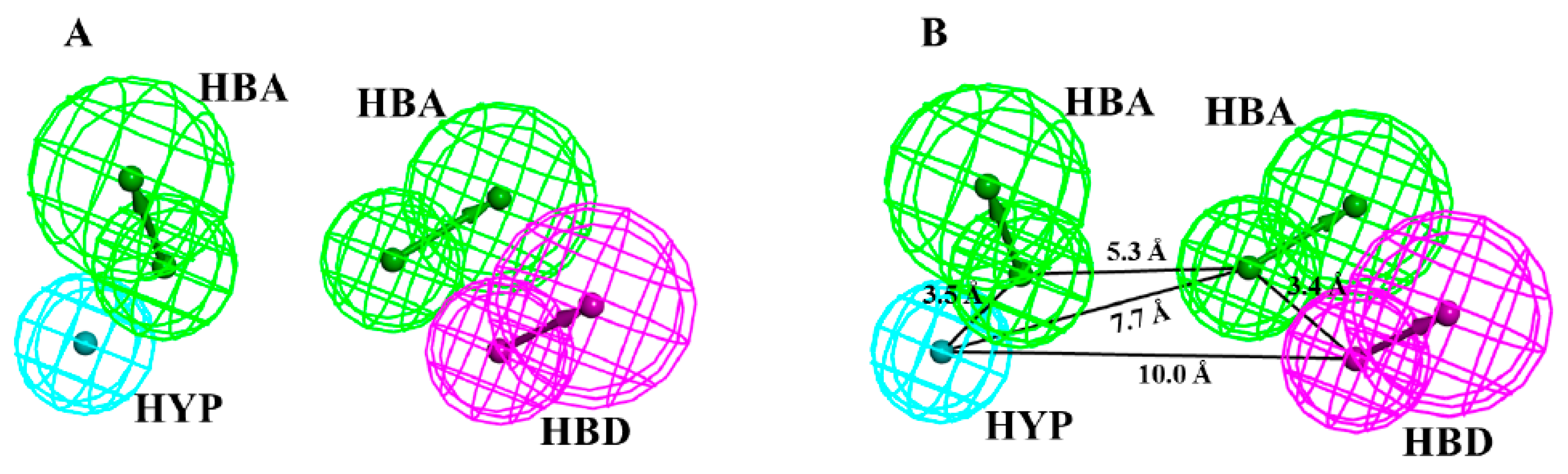
3.2. Quality Assessment Test (Validation) of Pharmacophore (Hypo1)
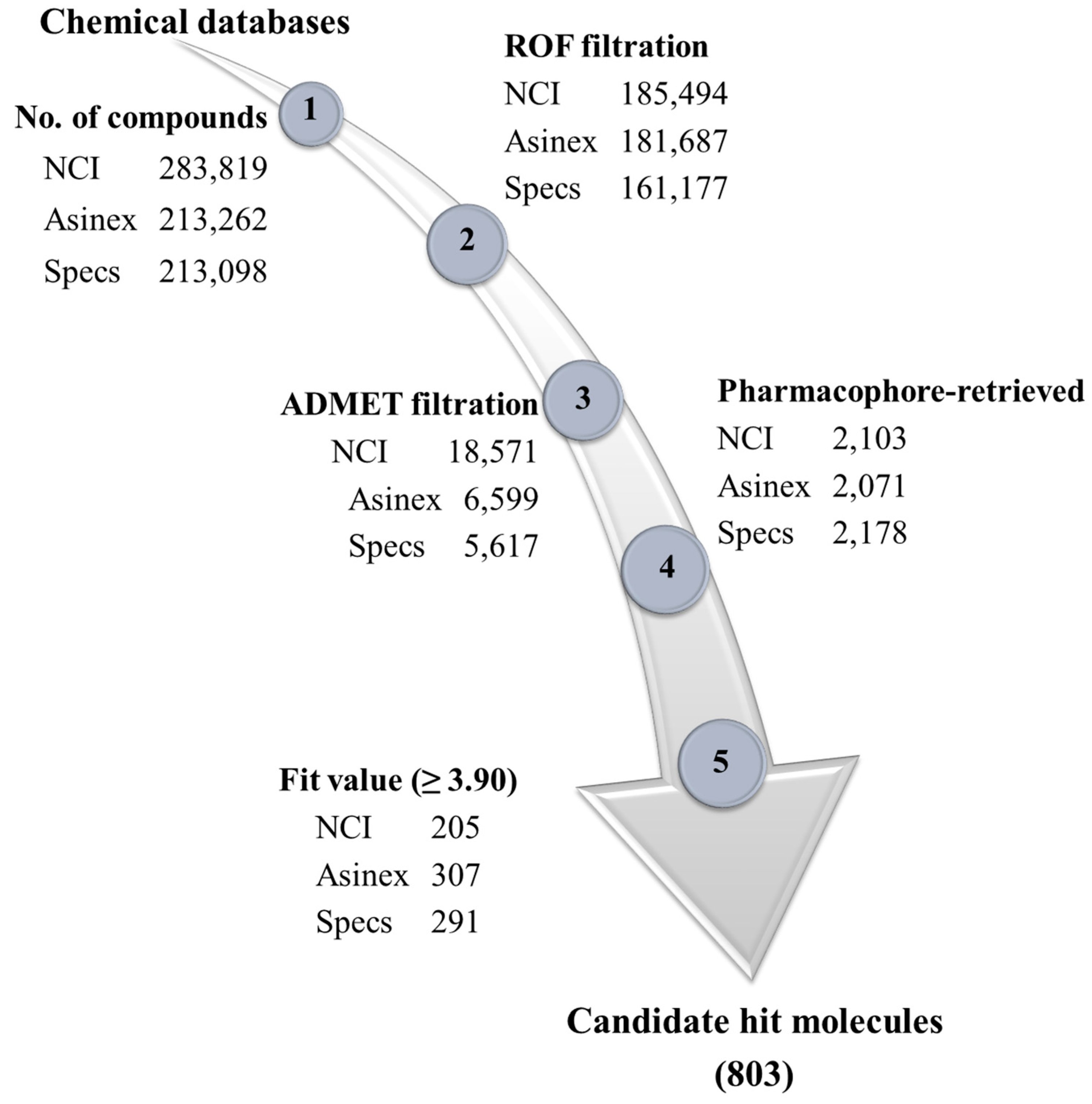
3.3. Development of Drug-like Database and Virtual Screening
3.4. Molecular Docking Simulation
3.5. Molecular Dynamics Simulation
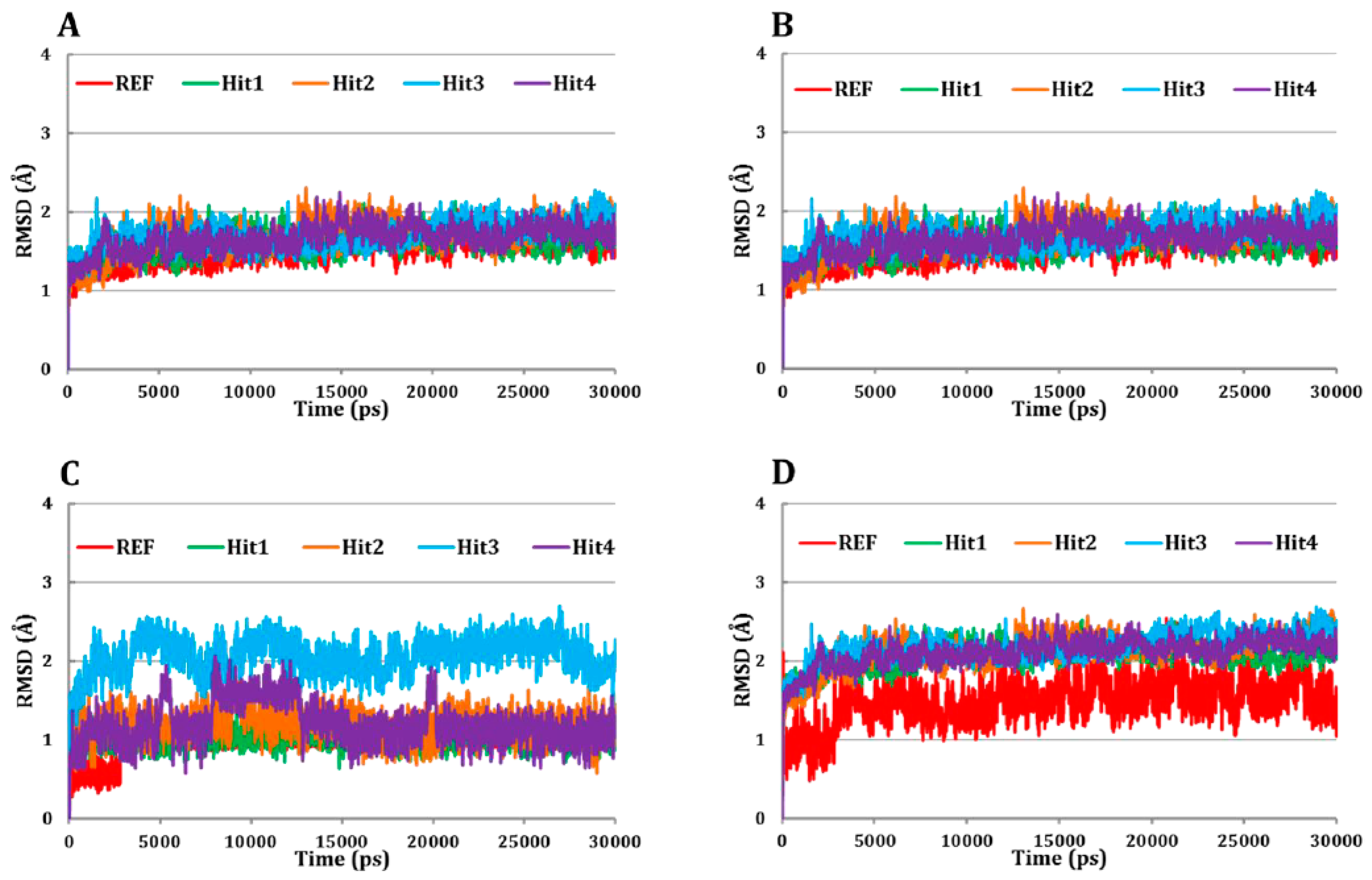
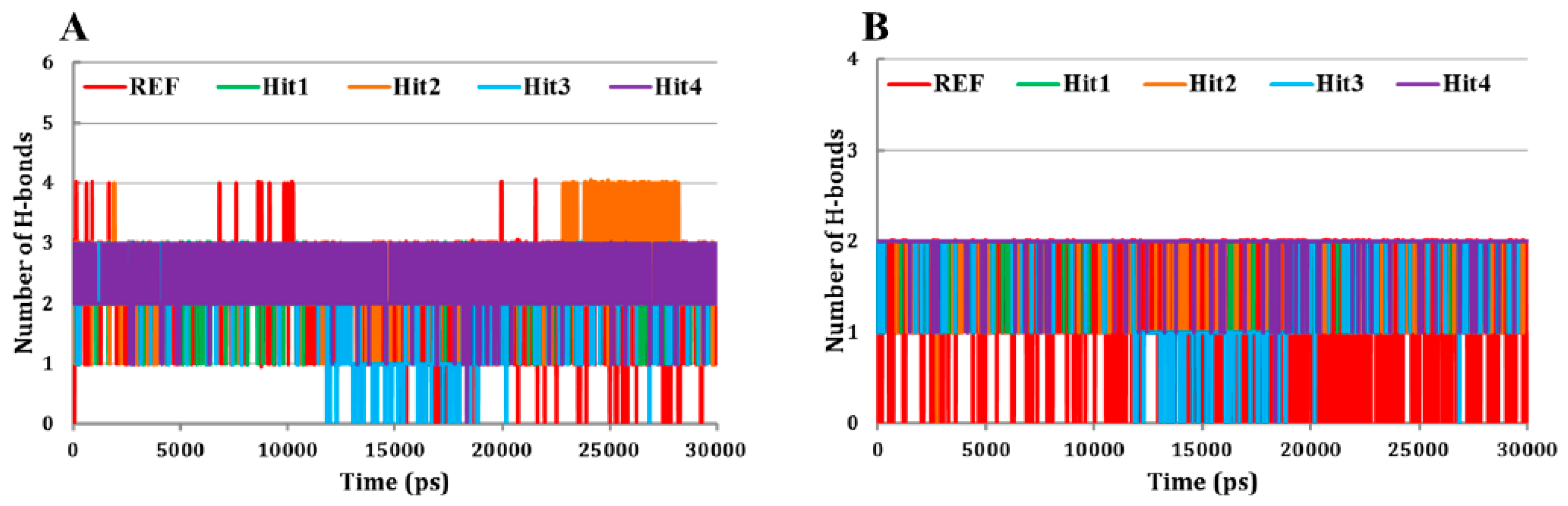
3.5.1. Root Mean Square Deviation (RMSD) Analysis
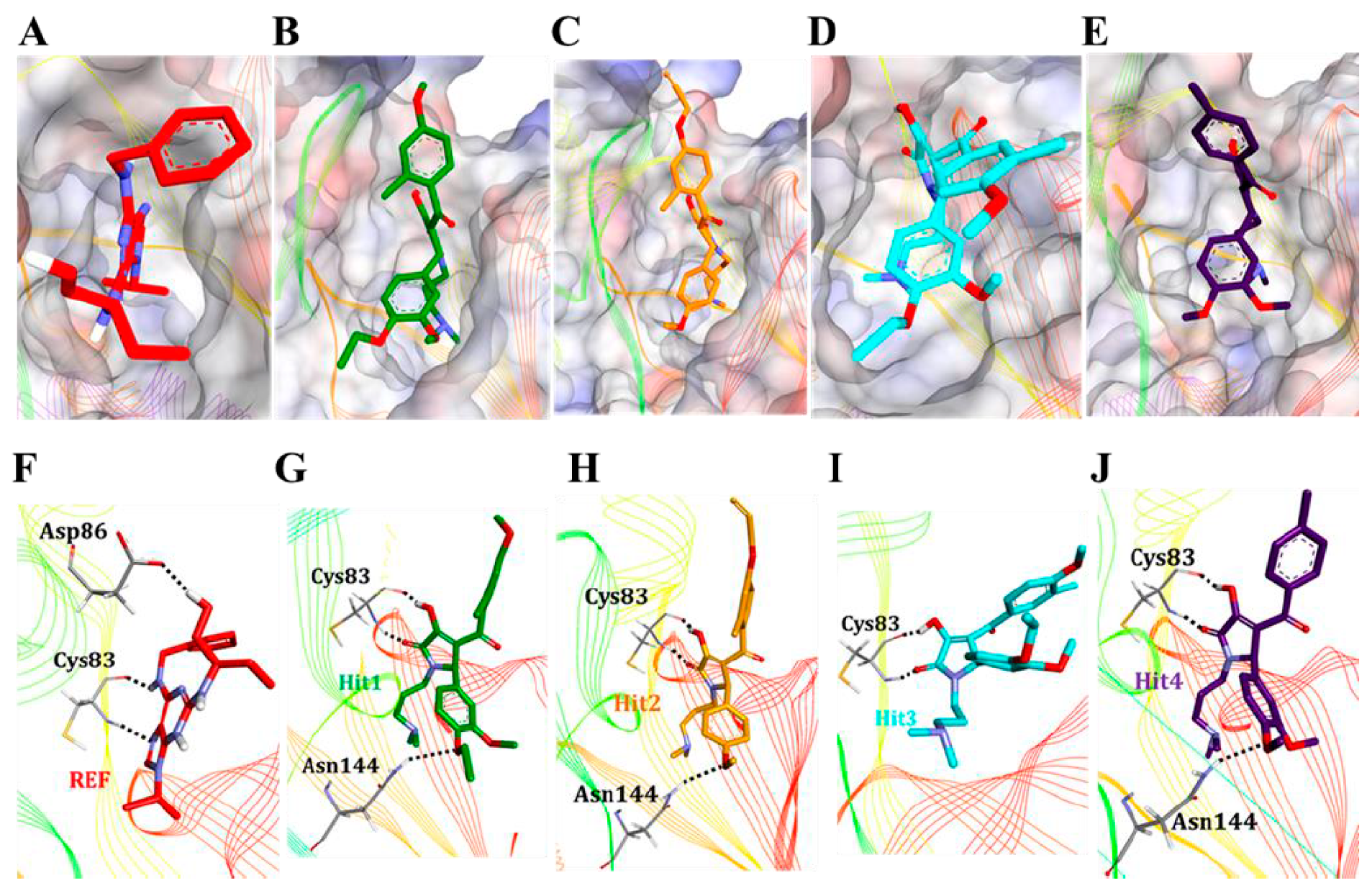
3.5.2. Molecular Overlay and Molecular Interaction Analysis
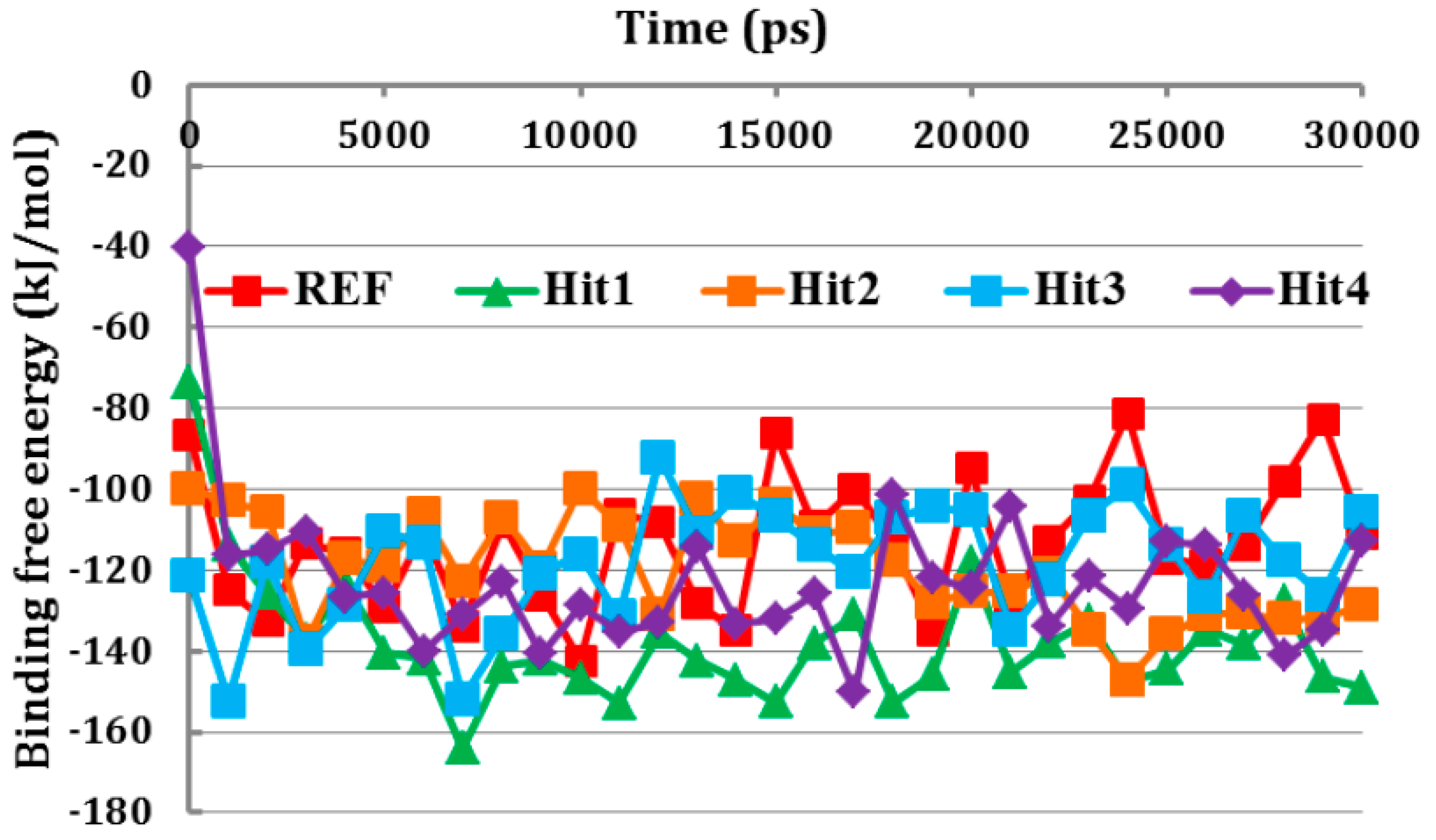
3.6. Binding Free Energy Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar]
- Hellmich, M.R.; Pant, H.C.; Wada, E.; Battey, J.F. Neuronal cdc2-like kinase: a cdc2-related protein kinase with predominantly neuronal expression. Proc. Natl. Acad. Sci. USA 1992, 89, 10867–10871. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Enders, G.H.; Wu, C.L.; Su, L.K.; Gorka, C.; Nelson, C.; Harlow, E.; Tsai, L.H. A family of human cdc2-related protein kinases. EMBO J. 1992, 11, 2909–2917. [Google Scholar]
- Tsai, L.H.; Takahashi, T.; Caviness, V.S.; Harlow, E. Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development 1993, 119, 1029–1040. [Google Scholar] [PubMed]
- Lew, J.; Beaudette, K.; Litwin, C.M.; Wang, J.H. Purification and characterization of a novel proline-directed protein kinase from bovine brain. J. Biol. Chem. 1992, 267, 13383–13390. [Google Scholar] [PubMed]
- Brinkkoetter, P.T.; Olivier, P.; Wu, J.S.; Henderson, S.; Krofft, R.D.; Pippin, J.W.; Hockenbery, D.; Roberts, J.M.; Shankland, S.J. Cyclin I activates Cdk5 and regulates expression of Bcl-2 and Bcl-XL in postmitotic mouse cells. J. Clin. Investig. 2009, 119, 3089–3101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Tsai, L.-H. Cyclin-dependent kinase 5 and neuronal migration in the neocortex. Neurosignals 2003, 12, 173–179. [Google Scholar] [PubMed]
- Angelo, M.; Plattner, F.; Giese, K.P. Cyclin-dependent kinase 5 in synaptic plasticity, learning and memory. J. Neurochem. 2006, 99, 353–370. [Google Scholar] [PubMed]
- Hawasli, A.H.; Benavides, D.R.; Nguyen, C.; Kansy, J.W.; Hayashi, K.; Chambon, P.; Greengard, P.; Powell, C.M.; Cooper, D.C.; Bibb, J.A. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat. Neurosci. 2007, 10, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Ward, J.M.; Huh, C.G.; Longenecker, G.; Veeranna; Pant, H.C.; Brady, R.O.; Martin, L.J.; Kulkarni, A.B. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc. Natl. Acad. Sci. USA 1996, 93, 11173–11178. [Google Scholar] [CrossRef]
- Morabito, M.A.; Sheng, M.; Tsai, L.-H. Cyclin-dependent kinase 5 phosphorylates the N-terminal domain of the postsynaptic density protein PSD-95 in neurons. J. Neurosci. 2004, 24, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Hawasli, A.H.; Koovakkattu, D.; Hayashi, K.; Anderson, A.E.; Powell, C.M.; Sinton, C.M.; Bibb, J.A.; Cooper, D.C. Regulation of hippocampal and behavioral excitability by cyclin-dependent kinase 5. PLoS ONE 2009, 4, e5808. [Google Scholar] [CrossRef]
- Lau, L.-F.; Ahlijanian, M.K. Role of cdk5 in the pathogenesis of Alzheimer’s disease. Neurosignals 2003, 12, 209–214. [Google Scholar] [CrossRef]
- Nguyen, M.D.; Julien, J.-P. Cyclin-dependent kinase 5 in Amyotrophic Lateral Sclerosis. Neurosignals 2003, 12, 215–220. [Google Scholar] [CrossRef]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.; Mandelkow, E.M. Microtubules and microtubule-associated proteins. Curr. Opin. Cell Biol. 1995, 7, 72–81. [Google Scholar] [CrossRef]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.-L.; Teng, H.-J.; Yin, B.; Xu, Y.; Du, Y.; He, F.-P.; Chu, K.-T.; Luo, B.-Y.; Zheng, G.-Q. A systematic review and meta-analysis of buyang huanwu decoction in animal model of focal cerebral ischemia. Evid. Based Complement. Altern. Med. 2013, 2013, 138484. [Google Scholar] [CrossRef]
- Cruz, J.C.; Tseng, H.-C.; Goldman, J.A.; Shih, H.; Tsai, L.-H. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 2003, 40, 471–483. [Google Scholar] [CrossRef]
- Rao, M.V.; McBrayer, M.K.; Campbell, J.; Kumar, A.; Hashim, A.; Sershen, H.; Stavrides, P.H.; Ohno, M.; Hutton, M.; Nixon, R.A. Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. J. Neurosci. 2014, 34, 9222–9234. [Google Scholar] [CrossRef]
- Bajaj, N.P. Cyclin-dependent kinase-5 (CDK5) and amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 319–327. [Google Scholar] [CrossRef]
- Patzke, H.; Tsai, L.H. Cdk5 sinks into ALS. Trends Neurosci. 2002, 25, 8–10. [Google Scholar] [CrossRef]
- Nguyen, M.D.; Larivière, R.C.; Julien, J.P. Deregulation of Cdk5 in a mouse model of ALS: Toxicity alleviated by perikaryal neurofilament inclusions. Neuron 2001, 30, 135–147. [Google Scholar] [CrossRef]
- Piedrahita, D.; Hernandez, I.; Lopez-Tobon, A.; Fedorov, D.; Obara, B.; Manjunath, B.S.; Boudreau, R.L.; Davidson, B.; LaFerla, F.; Gallego-Gomez, J.C.; et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer’s mice. J. Neurosci. 2010, 30, 13966–13976. [Google Scholar] [CrossRef]
- Plattner, F.; Hernández, A.; Kistler, T.M.; Pozo, K.; Zhong, P.; Yuen, E.Y.; Tan, C.; Hawasli, A.H.; Cooke, S.F.; Nishi, A.; et al. Memory enhancement by targeting Cdk5 regulation of NR2B. Neuron 2014, 81, 1070–1083. [Google Scholar] [CrossRef]
- Chang, K.-H.; Vincent, F.; Shah, K. Deregulated Cdk5 triggers aberrant activation of cell cycle kinases and phosphatases inducing neuronal death. J. Cell Sci. 2012, 125, 5124–5137. [Google Scholar] [CrossRef]
- Barnett, D.G.S.; Bibb, J.A. The role of Cdk5 in cognition and neuropsychiatric and neurological pathology. Brain Res. Bull. 2010, 85, 9–13. [Google Scholar] [CrossRef]
- Tan, X.; Chen, Y.; Li, J.; Li, X.; Miao, Z.; Xin, N.; Zhu, J.; Ge, W.; Feng, Y.; Xu, X. The inhibition of Cdk5 activity after Hypoxia/Ischemia injury reduces infract size and promotes functional recovery in neonatal rats. Neuroscience 2015, 290, 552–560. [Google Scholar] [CrossRef]
- Gillardon, F.; Schrattenholz, A.; Sommer, B. Investigating the neuroprotective mechanism of action of a CDK5 inhibitor by phosphoproteome analysis. J. Cell. Biochem. 2005, 95, 817–826. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, C.; Lü, J.; Xie, M.; Pan, D.; Luo, X.; Yu, Z.; Dong, Q.; Wang, W. Cell cycle inhibition attenuates microglial proliferation and production of IL-1β, MIP-1α, and NO after focal cerebral ischemia in the rat. Glia 2009, 57, 908–920. [Google Scholar] [CrossRef]
- Mushtaq, G.; Greig, N.H.; Anwar, F.; Al-Abbasi, F.A.; Zamzami, M.A.; Al-Talhi, H.A.; Kamal, M.A. Neuroprotective mechanisms mediated by CDK5 inhibition. Curr. Pharm. Des. 2016, 22, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Mapelli, M.; Massimiliano, L.; Crovace, C.; Seeliger, M.A.; Tsai, L.-H.; Meijer, L.; Musacchio, A. Mechanism of Cdk5/p25 binding by Cdk inhibitors. J. Med. Chem. 2005, 48, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Malmström, J.; Viklund, J.; Slivo, C.; Costa, A.; Maudet, M.; Sandelin, C.; Hiller, G.; Olsson, L.-L.; Aagaard, A.; Geschwindner, S.; et al. Synthesis and structure-activity relationship of 4-(1,3-benzothiazol-2-yl)-thiophene-2-sulfonamides as cyclin-dependent kinase 5 (cdk5)/p25 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5919–5923. [Google Scholar] [CrossRef]
- Schonbrunn, E.; Betzi, S.; Alam, R.; Martin, M.P.; Becker, A.; Han, H.; Francis, R.; Chakrasali, R.; Jakkaraj, S.; Kazi, A.; et al. Development of highly potent and selective diaminothiazole inhibitors of cyclin-dependent kinases. J. Med. Chem. 2013, 56, 3768–3782. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA, BIOVIA Workbook, Release 2017; BIOVIA Pipeline Pilot, Release 2017. Dassault Systèmes: San Diego, CA, USA. Available online: http://www.3dsbiovia.com/ (accessed on 4 December 2018).
- Jonesa, G.; Willett, P.; Glenb, R.C. A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. J. Comput. Aided Mol. Des. 1995, 9, 532–549. [Google Scholar] [CrossRef]
- Patil, S.; Tyagi, A.; Jose, J.; Menon, K.N.; Mohan, C.G. Integration of common feature pharmacophore modeling and in vitro study to identify potent AChE inhibitors. Med. Chem. Res. 2016, 25, 2965–2975. [Google Scholar] [CrossRef]
- Patel, C.N.; Georrge, J.J.; Modi, K.M.; Narechania, M.B.; Patel, D.P.; Gonzalez, F.J.; Pandya, H.A. Pharmacophore-based virtual screening of catechol-o-methyltransferase (COMT) inhibitors to combat Alzheimer’s disease. J. Biomol. Struct. Dyn. 2017, 1–20. [Google Scholar] [CrossRef]
- Zeb, A.; Park, C.; Rampogu, S.; Son, M.; Lee, G.; Lee, K.W. Structure-based drug designing recommends HDAC6 inhibitors to attenuate microtubule-associated Tau-pathogenesis. ACS Chem. Neurosci. 2018, 10, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development q settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Zeb, A.; Park, C.; Son, M.; Baek, A.; Cho, Y.; Kim, D.; Rampogu, S.; Lee, G.; Kwak, Y.-S.; Park, S.J.; et al. Integration of virtual screening and computational simulation identifies photodynamic therapeutics against human Protoporphyrinogen Oxidase IX (hPPO). Arab. J. Chem. 2018. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Guariento, S.; Bruno, O.; Fossa, P.; Cichero, E. New insights into PDE4B inhibitor selectivity: CoMFA analyses and molecular docking studies. Mol. Divers. 2016, 20, 77–92. [Google Scholar] [CrossRef]
- Franchini, S.; Manasieva, L.I.; Sorbi, C.; Battisti, U.M.; Fossa, P.; Cichero, E.; Denora, N.; Iacobazzi, R.M.; Cilia, A.; Pirona, L.; et al. Synthesis, biological evaluation and molecular modeling of 1-oxa-4-thiaspiro- and 1,4-dithiaspiro[4.5]decane derivatives as potent and selective 5-HT1A receptor agonists. Eur. J. Med. Chem. 2017, 125, 435–452. [Google Scholar] [CrossRef] [Green Version]
- Guariento, S.; Tonelli, M.; Espinoza, S.; Gerasimov, A.S.; Gainetdinov, R.R.; Cichero, E. Rational design, chemical synthesis and biological evaluation of novel biguanides exploring species-specificity responsiveness of TAAR1 agonists. Eur. J. Med. Chem. 2018, 146, 171–184. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Med. Chem. 1997, 56, 3768–3782. [Google Scholar]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation Methods and General Results. J. Chem. Inf. Model. 2014, 54, 1717–1736. [Google Scholar] [CrossRef]
- Mooij, W.T.M.; Verdonk, M.L. General and targeted statistical potentials for protein-ligand interactions. Proteins Struct. Funct. Bioinform. 2005, 61, 272–287. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Sakkiah, S.; Thangapandian, S.; John, S.; Kwon, Y.J.; Lee, K.W. 3D QSAR pharmacophore based virtual screening and molecular docking for identification of potential HSP90 inhibitors. Eur. J. Med. Chem. 2010, 45, 2132–2140. [Google Scholar] [CrossRef]
- Yao, T.-T.; Xie, J.-F.; Liu, X.-G.; Cheng, J.-L.; Zhu, C.-Y.; Zhao, J.-H.; Dong, X.-W. Integration of pharmacophore mapping and molecular docking in sequential virtual screening: towards the discovery of novel JAK2 inhibitors. RSC Adv. 2017, 7, 10353–10360. [Google Scholar] [CrossRef] [Green Version]
- Rampogu, S.; Baek, A.; Zeb, A.; Lee, K.W. Exploration for novel inhibitors showing back-to-front approach against VEGFR-2 kinase domain (4AG8) employing molecular docking mechanism and molecular dynamics simulations. BMC Cancer 2018, 18, 264. [Google Scholar] [CrossRef]
- Ravikumar, M.; Pavan, S.; Bairy, S.; Pramod, A.B.; Sumakanth, M.; Kishore, M.; Sumithra, T. Virtual screening of Cathepsin K inhibitors using docking and pharmacophore models. Chem. Biol. Drug Des. 2008, 72, 79–90. [Google Scholar] [CrossRef]
- Franchini, S.; Battisti, U.M.; Prandi, A.; Tait, A.; Borsari, C.; Cichero, E.; Fossa, P.; Cilia, A.; Prezzavento, O.; Ronsisvalle, S.; et al. Scouting new sigma receptor ligands: Synthesis, pharmacological evaluation and molecular modeling of 1,3-dioxolane-based structures and derivatives. Eur. J. Med. Chem. 2016, 112, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.S.; Radhakrishnan, M.L.; Mapelli, M.; Choi, S.; Tidor, B.; Cuny, G.D.; Musacchio, A.; Yeh, L.-A.; Kosik, K.S. Defining Cdk5 ligand chemical space with small molecule inhibitors of Tau phosphorylation. Chem. Biol. 2005, 12, 811–823. [Google Scholar] [CrossRef]
- Sahu, V.K.; Khan, A.K.R.; Singh, R.K.; Singh, P.P. Hydrophobic, polar and hydrogen bonding based drug-receptor interaction of tetrahydroimidazobenzodiazepinones. Am. J. Immunol. 2008, 4, 33–42. [Google Scholar] [CrossRef]
- Patel, S.K.; Lavasanifar, A.; Choi, P. Roles of nonpolar and polar intermolecular interactions in the improvement of the drug loading capacity of PEO-b-PCL with increasing PCL content for two hydrophobic cucurbitacin drugs. Biomacromolecules 2009, 10, 2584–2591. [Google Scholar] [CrossRef]
- Rath, S.L.; Senapati, S. Molecular basis of differential selectivity of cyclobutyl-substituted imidazole inhibitors against CDKs: Insights for rational drug design. PLoS ONE 2013, 8, 73836. [Google Scholar] [CrossRef]
- Hamdouchi, C.; Keyser, H.; Collins, E.; Jaramillo, C.; De Diego, J.E.; Spencer, C.D.; Dempsey, J.A.; Anderson, B.D.; Leggett, T.; Stamm, N.B.; et al. The discovery of a new structural class of cyclin-dependent kinase inhibitors, aminoimidazo[1,2-a]pyridines. Mol. Cancer Ther. 2004, 3, 1–9. [Google Scholar]
- Bettayeb, K.; Baunbæk, D.; Delehouze, C.; Loaëc, N.; Hole, A.J.; Baumli, S.; Endicott, J.A.; Douc-Rasy, S.; Bénard, J.; Oumata, N.; et al. CDK inhibitors roscovitine and CR8 trigger Mcl-1 down-regulation and apoptotic cell death in neuroblastoma cells. Genes Cancer 2010, 1, 369–380. [Google Scholar] [CrossRef]
- Hole, A.J.; Baumli, S.; Shao, H.; Shi, S.; Huang, S.; Pepper, C.; Fischer, P.M.; Wang, S.; Endicott, J.A.; Noble, M.E. Comparative structural and functional studies of 4-(thiazol-5-yl)-2-(phenylamino)pyrimidine-5-carbonitrile CDK9 inhibitors suggest the basis for isotype selectivity. J. Med. Chem. 2013, 56, 660–670. [Google Scholar] [CrossRef]
- Brown, N.R.; Korolchuk, S.; Martin, M.P.; Stanley, W.A.; Moukhametzianov, R.; Noble, M.E.M.; Endicott, J.A. CDK1 structures reveal conserved and unique features of the essential cell cycle CDK. Nat. Commun. 2015, 6, 6769. [Google Scholar] [CrossRef]
- Bergeron, P.; T Koehler, M.F.; Blackwood, E.M.; Bowman, K.; Clark, K.; Firestein, R.; Kiefer, J.R.; Maskos, K.; McCleland, M.L.; Orren, L.; et al. Design and development of a series of potent and selective type II inhibitors of CDK8. ACS Med. Chem. Lett. 2016, 7, 595–600. [Google Scholar] [CrossRef]
- Sarkar, A.; Kellogg, G. Hydrophobicity - Shake flasks, protein folding and drug discovery. Curr. Top. Med. Chem. 2010, 10, 67–83. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hypo. No. | Features a | Rank b | Direct Hit c | Partial Hit d | Max. Fit |
|---|---|---|---|---|---|
| 1 | HYP, HBD, HBA, HBA | 75.175 | 11111111 | 00000000 | 4 |
| 2 | HYP, HBD, HBA, HBA | 74.277 | 11111111 | 00000000 | 4 |
| 3 | HYP, HBD, HBA, HBA | 70.057 | 11111111 | 00000000 | 4 |
| 4 | HYP, HBD, HBA, HBA | 68.758 | 11111111 | 00000000 | 4 |
| 5 | HYP, HYP, HBD, HBA | 64.341 | 11111111 | 00000000 | 4 |
| 6 | HYP, HYP, HBD, HBA | 63.638 | 11111111 | 00000000 | 4 |
| 7 | HYP, HYP, HBD, HBA | 62.807 | 11111111 | 00000000 | 4 |
| 8 | HYP, HBD, HBA, HBA | 61.279 | 11111111 | 00000000 | 4 |
| 9 | HYP, HBA, HBD | 57.531 | 11111111 | 00000000 | 4 |
| 10 | HYP, HBD, HBA | 55.820 | 11111111 | 00000000 | 4 |
| S. No. | Parameter | Calculated Value |
|---|---|---|
| 1 | Total number of molecules in the database (D) | 708 |
| 2 | Total number of active molecules of Cdk5 in the database (A) | 40 |
| 3 | Total number of active molecules of Cdk5 in the retrieved hits (Ha) | 38 |
| 4 | Number of retrieved hits by pharmacophore (Ht) | 46 |
| 5 | % Yield of actives ((Ha/Ht) × 100] | 82.6 |
| 6 | % Ratio of actives ((Ha/A) × 100] | 95.0 |
| 7 | False positive (Ht − Ha) | 8 |
| 8 | False negative (A − Ha) | 2 |
| 9 | Goodness of fit (GH) | 0.84 |
| 10 | Enrichment factor (EF) | 14.62 |
| Compound | Docking Score | RMSD (Å) | Binding Free Energy (kJ/mol) | ||||
|---|---|---|---|---|---|---|---|
| Goldscore # | ASP $ | Complex | Cα Atoms ψ | Backbone Atoms | Inhibitors | ||
| REF | 67.67 | 26.32 | 1.54 | 1.54 | 1.54 | 1.02 | −113.10 |
| Hit1 | 79.22 | 33.14 | 2.04 | 1.59 | 1.57 | 1.02 | −137.90 |
| Hit2 | 77.39 | 30.64 | 2.12 | 1.70 | 1.68 | 1.20 | −119.65 |
| Hit3 | 86.33 | 35.31 | 2.18 | 1.70 | 1.68 | 2.06 | −118.22 |
| Hit4 | 75.35 | 30.29 | 2.10 | 1.65 | 1.63 | 1.20 | −122.65 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeb, A.; Kim, D.; Alam, S.I.; Son, M.; Kumar, R.; Rampogu, S.; Parameswaran, S.; Shelake, R.M.; Rana, R.M.; Parate, S.; et al. Computational Simulations Identify Pyrrolidine-2,3-Dione Derivatives as Novel Inhibitors of Cdk5/p25 Complex to Attenuate Alzheimer’s Pathology. J. Clin. Med. 2019, 8, 746. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050746
Zeb A, Kim D, Alam SI, Son M, Kumar R, Rampogu S, Parameswaran S, Shelake RM, Rana RM, Parate S, et al. Computational Simulations Identify Pyrrolidine-2,3-Dione Derivatives as Novel Inhibitors of Cdk5/p25 Complex to Attenuate Alzheimer’s Pathology. Journal of Clinical Medicine. 2019; 8(5):746. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050746
Chicago/Turabian StyleZeb, Amir, Donghwan Kim, Sayed Ibrar Alam, Minky Son, Raj Kumar, Shailima Rampogu, Saravanan Parameswaran, Rahul Mahadev Shelake, Rabia Mukhtar Rana, Shraddha Parate, and et al. 2019. "Computational Simulations Identify Pyrrolidine-2,3-Dione Derivatives as Novel Inhibitors of Cdk5/p25 Complex to Attenuate Alzheimer’s Pathology" Journal of Clinical Medicine 8, no. 5: 746. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050746