HCMV Infection in a Mesenchymal Stem Cell Niche: Differential Impact on the Development of NK Cells versus ILC3

 , , and
, , and
Abstract
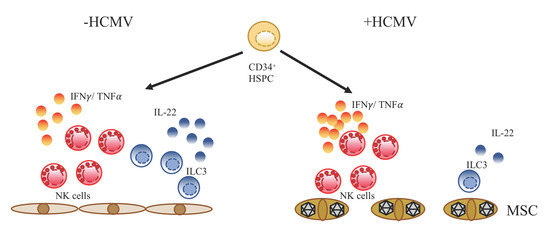
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Human Samples, Cell lines and Cell Culture
2.2. Infection of MSC with HCMV
2.3. Co-culturing MSCs/ 721.211 and HSCP
2.4. Flow Cytometric Analyses
2.5. Analysis of NK Cells and ILC3 Function
2.6. Statistical Analyses
3. Results
3.1. HCMV Infection and Virus Replication in Bone Marrow-Derived Human MSC
3.2. NK Cell Differentiation in the Presence of HCMV-Infected MSC
3.3. HCMV Infection Leads to Enhancement of NK Cell Effector Functions
3.4. Differential Impact of HCMV Infection on the Development of NK Cells versus ILC3
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frenette, P.S.; Pinho, S.; Lucas, D.; Scheiermann, C. Mesenchymal stem cell: Keystone of the Hematopoietic stem cell niche and a stepping-stone for regenerative medicine. Annu. Rev. Immunol. 2013, 31, 285–316. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, J.; Sensébé, L. Mesenchymal stromal cells: Clinical challenges and therapeutic opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, V.; Sciumè, G.; Santoni, A.; Bernardini, G. Bone marrow NK Cells: Origin, distinctive features; requirements for tissue localization. Front. Immunol. 2019, 10, 1569. [Google Scholar] [CrossRef] [Green Version]
- Freud, A.G.; Yokohama, A.; Becknell, B.; Lee, M.T.; Mao, H.C.; Ferketich, A.K.; Caligiuri, M.A. Evidence for discrete stages of human natural killer cell differentiation in vivo. J. Exp. Med. 2006, 203, 1033–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljunggren, H.-G.; Kärre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Manser, A.R.; Weinhold, S.; Uhrberg, M. Human KIR repertoires: Shaped by genetic diversity and evolution. Immunol. Rev. 2015, 267, 178–196. [Google Scholar] [CrossRef]
- Long, E.O.; Kim, H.S.; Liu, D.; Peterson, M.E.; Rajagopalan, S. Controlling natural killer cell responses: Integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [Green Version]
- Smyth, M.J.; Cretney, E.; Kelly, J.M.; Westwood, J.A.; Street, S.E.A.; Yagita, H.; Takeda, K.; van Dommelen, S.L.H.; Degli-Esposti, M.A.; Hayakawa, Y. Activation of NK cell cytotoxicity. Mol. Immunol. 2005, 42, 501–510. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells: 10 years on. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Mjösberg, J.M.; Trifari, S.; Crellin, N.K.; Peters, C.P.; van Drunen, C.M.; Piet, B.; Fokkens, W.J.; Cupedo, T.; Spits, H. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 2011, 12, 1055. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.M.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human NK cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Hoorweg, K.; Peters, C.P.; Cornelissen, F.; Aparicio-Domingo, P.; Papazian, N.; Kazemier, G.; Mjösberg, J.M.; Spits, H.; Cupedo, T. Functional differences between human NKp44(−) and NKp44(+) RORC(+) innate lymphoid cells. Front. Immunol. 2012, 3, 72. [Google Scholar] [CrossRef] [Green Version]
- Renoux, V.M.; Zriwil, A.; Peitzsch, C.; Michaëlsson, J.; Friberg, D.; Soneji, S.; Sitnicka, E. Identification of a human natural killer cell lineage-restricted progenitor in fetal and adult tissues. Immunity 2015, 43, 394–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoville, S.D.; Mundy-Bosse, B.L; Zhang, M.H.; Chen, L.; Zhang, X.; Keller, K.A.; Hughes, T.; Chen, L.; Cheng, S.; Bergin, S.M.; et al. A progenitor cell expressing transcription factor RORγt generates all human innate lymphoid cell subsets. Immunity 2016, 44, 1140–1150. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Cherrier, D.E.; Chea, S.; Vosshenrich, C.; Serafini, N.; Petit, M.; Liu, P.; Golub, R.; Di Santo, J.P. An Id2(RFP)-reporter mouse redefines innate lymphoid cell precursor potentials. Immunity 2019, 50, 1054–1068. [Google Scholar] [CrossRef] [Green Version]
- Brodin, P.; Jojic, V.; Gao, T.; Bhattacharya, S.; Angel, C.J.L.; Furman, D.; Shen-Orr, S.; Dekker, C.L.; Swan, G.E.; Butte, A.J.; et al. Variation in the human immune system is largely driven by non-heritable influences. Cell 2015, 160, 37–47. [Google Scholar] [CrossRef] [Green Version]
- López-Botet, M.; Muntasell, A.; Vilches, C. The CD94/NKG2C+ NK-cell subset on the edge of innate and adaptive immunity to human cytomegalovirus infection. Semin. Immunol. 2014, 26, 145–151. [Google Scholar] [CrossRef]
- Béziat, V.; Liu, L.L.; Malmberg, J.-A.; Ivarsson, M.A.; Sohlberg, E.; Björklund, A.T.; Retière, C.; Sverremark-Ekström, E.; Traherne, J.; Ljungman, P.; et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 2013, 121, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Manser, A.R.; Scherenschlich, N.; Thöns, C.; Hengel, H.; Timm, J.; Uhrberg, M. KIR polymorphism modulates the size of the adaptive NK cell pool in human cytomegalovirus–infected individuals. J. Immunol. 2019, 203, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S. Immunomodulation by cytomegaloviruses: Manipulative strategies beyond evasion. Trends Microbiol. 2002, 10, 332–339. [Google Scholar] [CrossRef]
- Landolfo, S.; Gariglio, M.; Gribaudo, G.; Lembo, D. The human cytomegalovirus. Pharmacol. Ther. 2003, 98, 269–297. [Google Scholar] [CrossRef]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Cooley, S.; Davis, Z.; DeFor, T.E.; Schlums, H.; Zhang, B.; Brunstein, C.G.; Blazar, B.R.; Wagner, J.; Diamond, D.J.; et al. CD56dimCD57 + NKG2C + NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia 2016, 30, 456–463. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Falco, M.; Muccio, L.; Bertaina, A.; Locatelli, F.; Moretta, A. Impact of HCMV infection on NK cell development and function after HSCT. Front. Immunol. 2013, 4, 458. [Google Scholar] [CrossRef] [Green Version]
- Della Chiesa, M.; Muccio, L.; Moretta, A. CMV induces rapid NK cell maturation in HSCT recipients. Immunol. Lett. 2013, 155, 11–13. [Google Scholar] [CrossRef]
- Elmaagacli, A.H.; Steckel, N.K.; Koldehoff, M.; Hegerfeldt, Y.; Trenschel, R.; Ditschkowski, M.; Christoph, S.; Gromke, T.; Kordelas, L.; Ottinger, H.D.; et al. Early human cytomegalovirus replication after transplantation is associated with a decreased relapse risk: Evidence for a putative virus-versus-leukemia effect in acute myeloid leukemia patients. Blood 2011, 118, 1402–1412. [Google Scholar] [CrossRef] [Green Version]
- McCullar, V.; Oostendorp, R.; Panoskaltsis-Mortari, A.; Yun, G.; Lutz, C.T.; Wagner, J.E.; Miller, J.S. Mouse fetal and embryonic liver cells differentiate human umbilical cord blood progenitors into CD56-negative natural killer cell precursors in the absence of interleukin-15. Exp. Hematol. 2008, 36, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Weinhold, S.; Brands, J.; Hejazi, M.; Degistirici, Ö.; Kögler, G.; Meisel, R.; Uhrberg, M. NK cell development in a human stem cell niche: KIR expression occurs independently of the presence of HLA class I ligands. Blood Adv. 2018, 2, 2452–2461. [Google Scholar] [CrossRef] [PubMed]
- Meisel, R.; Zibert, A.; Laryea, M.; Gobel, U.; Däubener, W.; Dilloo, D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2, 3-dioxygenase–mediated tryptophan degradation. Blood 2004, 103, 4619–4621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najar, M.; Bouhtit, F.; Melki, R.; Afif, H.; Hamal, A.; Fahmi, H.; Merimi, M.; Lagneaux, L. Mesenchymal stromal cell-based therapy: New perspectives and challenges. J. Clin. Med. 2019, 8, 626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisel, R.; Heseler, K.; Nau, J.; Schmidt, S.K.; Leineweber, M.; Pudelko, S.; Wenning, J.; Zimmermann, A.; Hengel, H.; Sinzger, C.; et al. Cytomegalovirus infection impairs immunosuppressive and antimicrobial effector functions of human multipotent mesenchymal stromal cells. Mediat. Inflamm. 2014, 2014, 898630. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, S.V.; Harbacheuski, R.; Lewis-Antes, A.; Zhu, H.; Rameshwar, P.; Kotenko, S.V. Bone-marrow-derived mesenchymal stem cells as a target for cytomegalovirus infection: Implications for hematopoiesis, self-renewal and differentiation potential. Virology 2007, 360, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Britanova, L.; Diefenbach, A. Interplay of innate lymphoid cells and the microbiota. Immunol. Rev. 2017, 279, 36–51. [Google Scholar] [CrossRef]
- Hejazi, M.; Manser, A.R.; Fröbel, J.; Kündgen, A.; Zhao, X.; Schönberg, K.; Germing, U.; Haas, R.; Gattermann, N.; Uhrberg, M. Impaired cytotoxicity associated with defective natural killer cell differentiation in myelodysplastic syndromes. Haematologica 2015, 100, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Hesse, J.; Reyda, S.; Tenzer, S.; Besold, K.; Reuter, N.; Krauter, S.; Büscher, N.; Stamminger, T.; Plachter, B. Human cytomegalovirus pp71 stimulates major histocompatibility complex class i presentation of IE1-derived peptides at immediate early times of infection. J. Virol. 2013, 87, 5229–5238. [Google Scholar] [CrossRef] [Green Version]
- Wroblewska, Z.; Wellish, M.C.; Wolinsky, J.S.; Gilden, D. Comparison of human cytomegalovirus growth in MRC-5 human fibroblasts, brain; choroid plexus cells in vitro. J. Med. Virol. 1981, 8, 245–256. [Google Scholar] [CrossRef]
- Schönberg, K.; Fischer, J.C.; Kögler, G.; Uhrberg, M. Neonatal NK-cell repertoires are functionally, but not structurally, biased toward recognition of self HLA class I. Blood 2011, 117, 5152–5156. [Google Scholar] [CrossRef] [Green Version]
- Carson, W.E.; Haldar, S.; Baiocchi, R.A.; Croce, C.M.; Caligiuri, M.A. The c-kit ligand suppresses apoptosis of human natural killer cells through the upregulation of bcl-2. Proc. Natl. Acad. Sci. USA 1994, 91, 7553–7557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Uhrberg, M. The CD107 mobilization assay: Viable isolation and immunotherapeutic potential of tumor-cytolytic NK cells. Leukemia 2005, 19, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.P.; Martin, S.J. Mechanisms of granule-dependent killing. Cell Death Differ. 2008, 15, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Ahn, Y.-O.; Blazar, B.R.; Miller, J.S.; Verneris, M.R. Lineage relationships of human interleukin-22–producing CD56+ RORγt+ innate lymphoid cells and conventional natural killer cells. Blood 2013, 121, 2234–2243. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Naito, A.; Inoue, J.-I.; Satoh, M.; Santee-Cooper, S.M.; Ware, C.F.; Togawa, A.; Nishikawa, S.; Nishikawa, S.-I. Different cytokines induce surface lymphotoxin-αβ on IL-7 receptor-α cells that differentially engender lymph nodes and Peyer’s patches. Immunity 2002, 17, 823–833. [Google Scholar] [CrossRef] [Green Version]
- Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non–major histocompatibility complex–restricted tumor cell lysis. J. Exp. Med. 1998, 187, 2065–2072. [Google Scholar] [CrossRef]
- Hammer, Q.; Rückert, T.; Romagnani, C. Natural killer cell specificity for viral infections. Nat. Immunol. 2018, 19, 800–808. [Google Scholar] [CrossRef]
- Nabekura, T.; Lanier, L.L. Tracking the fate of antigen-specific versus cytokine-activated natural killer cells after cytomegalovirus infection. J. Exp. Med. 2016, 213, 2745–2758. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Ahn, Y.-O.; Southern, P.; Blazar, B.R.; Miller, J.S.; Verneris, M.R. Development of IL-22—Producing NK lineage cells from umbilical cord blood hematopoietic stem cells in the absence of secondary lymphoid tissue. Blood 2011, 117, 4052–4055. [Google Scholar] [CrossRef]
- van Hoeven, V.; Munneke, J.M.; Cornelissen, A.S.; Omar, S.Z.; Spruit, M.J.; Kleijer, M.; Bernink, J.H.; Blom, B.; Voermans, C.; Hazenberg, M.D. Mesenchymal stromal cells stimulate the proliferation and IL-22 production of group 3 innate lymphoid cells. J. Immunol. 2018, 201, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Bar-Ephraim, Y.E.; Cornelissen, F.; Papazian, N.; Konijn, T.; Hoogenboezem, R.M.; Sanders, M.A.; Westerman, B.A.; Gönültas, M.; Kwekkeboom, J.; Den Haan, J.M.M.; et al. Cross-tissue transcriptomic analysis of human secondary lymphoid organ-residing ILC3s reveals a quiescent state in the absence of inflammation. Cell Rep. 2017, 21, 823–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaldo, E.; Juelke, K.; Romagnani, C. Group 3 innate lymphoid cells (ILC3s): Origin, differentiation; plasticity in humans and mice. Eur. J. Immunol. 2015, 45, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.I.; Li, Y.; Lopez-Lastra, S.; Stadhouders, R.; Paul, F.; Casrouge, A.; Serafini, N.; Puel, A.; Bustamante, J.; Surace, L.; et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell 2017, 168, 1086–1100. [Google Scholar] [CrossRef] [Green Version]
- Mazzurana, L.; Rao, A.; Van Acker, A.; Mjösberg, J. The roles for innate lymphoid cells in the human immune system. Semin. Immunopathol. 2018, 40, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Geremia, A.; Arancibia-Cárcamo, C.V.; Fleming, M.P.P.; Rust, N.; Singh, B.; Mortensen, N.J.; Travis, S.P.L.; Powrie, F. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 2011, 208, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Takayama, T.; Kamada, N.; Chinen, H.; Okamoto, S.; Kitazume, M.T.; Chang, J.; Matuzaki, Y.; Suzuki, S.; Sugita, A.; Koganei, K.; et al. Imbalance of NKp44 + NKp46 − and NKp44 − NKp46 + Natural Killer Cells in the Intestinal mucosa of patients with Crohn’s disease. Gastroenterology 2010, 139, 882–892.e883. [Google Scholar] [CrossRef]
- Hanash, A.M.; Dudakov, J.A.; Hua, G.; O’Connor, M.H.; Young, L.F.; Singer, N.V.; West, M.L.; Jenq, R.R.; Holland, A.M.; Kappel, L.W.; et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft vs. host disease. Immunity 2012, 37, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Munneke, J.M.; Björklund, A.T.; Mjösberg, J.M.; Garming-Legert, K.; Bernink, J.H.; Blom, B.; van Oers, M.H.J.; Huisman, C.; Spits, H.; Malmberg, K-J.; et al. Activated innate lymphoid cells are associated with a reduced susceptibility to graft-versus-host disease. Blood 2014, 124, 812–821. [Google Scholar] [CrossRef]
- Cantoni, N.; Hirsch, H.H.; Khanna, N.; Gerull, S.; Buser, A.; Bucher, C.; Halter, J.; Heim, D.; Tichelli, A.; Gratwohl, A.; et al. Evidence for a bidirectional relationship between cytomegalovirus replication and acute graft-versus-host disease. Biol. Blood Marrow Transplant. 2010, 16, 1309–1314. [Google Scholar] [CrossRef] [Green Version]
- Kløverpris, H.N.; Kazer, S.W.; Mjösberg, J.; Mabuka, J.M.; Wellmann, A.; Ndhlovu, Z.; Yadon, M.C.; Nhamoyebonde, S.; Muenchhoff, M.; Simoni, Y.; et al. Innate lymphoid cells are depleted irreversibly during acute HIV-1 infection in the absence of viral suppression. Immunity 2016, 44, 391–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, B.; Goeser, F.; Lutz, P.; Glässner, A.; Boesecke, C.; Schwarze-Zander, C.; Kaczmarek, D.; Nischalke, H.D.; Branchi, V.; Manekeller, S.; et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathogens 2017, 13, e1006373. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, X.; Lackner, A.A.; Veazey, R.S. Type 3 innate lymphoid cell depletion is mediated by TLRs in lymphoid tissues of simian immunodeficiency virus–infected macaques. FASEB J. 2015, 29, 5072–5080. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ising, R.; Weinhold, S.; Bennstein, S.B.; Zimmermann, A.; Degistirici, Ö.; Kögler, G.; Meisel, R.; Hengel, H.; Timm, J.; Uhrberg, M. HCMV Infection in a Mesenchymal Stem Cell Niche: Differential Impact on the Development of NK Cells versus ILC3. J. Clin. Med. 2020, 9, 10. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9010010
Ising R, Weinhold S, Bennstein SB, Zimmermann A, Degistirici Ö, Kögler G, Meisel R, Hengel H, Timm J, Uhrberg M. HCMV Infection in a Mesenchymal Stem Cell Niche: Differential Impact on the Development of NK Cells versus ILC3. Journal of Clinical Medicine. 2020; 9(1):10. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9010010
Chicago/Turabian StyleIsing, Ricarda, Sandra Weinhold, Sabrina Bianca Bennstein, Albert Zimmermann, Özer Degistirici, Gesine Kögler, Roland Meisel, Hartmut Hengel, Jörg Timm, and Markus Uhrberg. 2020. "HCMV Infection in a Mesenchymal Stem Cell Niche: Differential Impact on the Development of NK Cells versus ILC3" Journal of Clinical Medicine 9, no. 1: 10. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9010010