Sequence Variation in the DDAH1 Gene Predisposes for Delayed Cerebral Ischemia in Subarachnoidal Hemorrhage

 ,
,
Abstract
:1. Introduction

2. Materials and Methods
2.1. Study Participants and Protocol
2.2. Clinical Assessment and Follow-up
2.3. Selection of SNPs
2.4. DNA Isolation and SNP Genotyping
2.5. Biochemical Analyses
2.6. Statistical Analyses
3. Results
3.1. Baseline Characteristics and Clinical Outcome of the Patients
3.2. Association of L-Arginine and Dimethylarginines with DCI
3.3. Allele Frequencies of Genes of Interest in the Study Cohort Compared to the European Reference Population
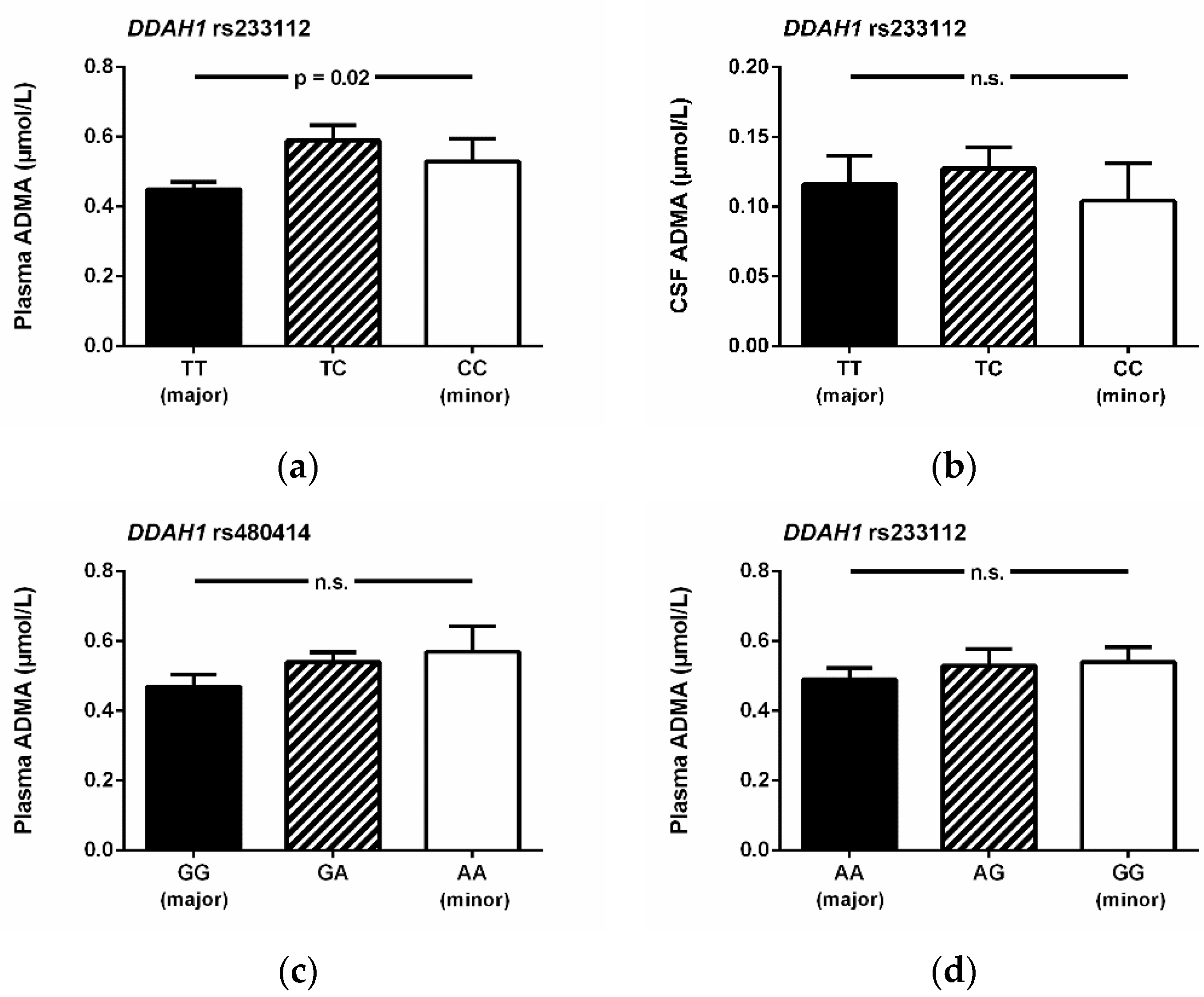
3.4. Association of SNPs with Biomarkers in Plasma and in Cerebrospinal Fluid
3.5. Association of SNPs with the Incidence of Delayed Cerebral Ischemia
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Selection and Clinical Significance of the Selected Single Nucleotide Polymorphisms
Appendix A.2. Construction of DDAH1 Haplotypes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/SNP | Position | Genetic Consequence | Clinical Rationale | Related References |
|---|---|---|---|---|
| NOS3 | ||||
| rs1799983 | Exon 8 | Missense variant | Minor allele associated with worse outcome after SAH and risk of ischemic stroke. | [26,49,50,51,52] |
| rs2070744 | Intron 1 | Intron variant | Minor allele associated with increased risk of DCI and cardiac instability in patients with SAH; proposed as a susceptibility locus for cerebral infarction. | [53,54,55] |
| rs891512 | Intron 23 | Intron variant | Minor allele associated with ankle-brachial index, with higher systolic and diastolic blood pressure, and risk of coronary artery disease. | [56,57,58,59,60] |
| DDAH1 | ||||
| rs1241321 | Intron 1 | Intron variant | Minor allele associated with higher risk of all-cause mortality and the combined endpoint of cardiovascular death, nonfatal myocardial infarction and stroke. | [61] |
| rs233112 | Exon 6 | 3′ UTR variant | Minor allele associated with arterial stiffness and pulse wave reflection. | [62] |
| rs480414 | Intron 1 | Intron variant | Minor allele associated with a lower incidence of pulmonary hypertension in patients with bronchopulmonary dysplasia. | [63] |
| DDAH2 | ||||
| rs805304 | 2 kb upstream variant | Minor allele associated with higher prevalence of hypertension, higher ADMA concentration in diabetic renal impairment, and decreased risk of myocardial infarction. | [64,65,66] | |
| rs2272592 | 2 kb upstream variant | Minor allele associated with type 2 diabetes. | [67] | |
| ARG1 | ||||
| rs2246012 | Intron 2 | Intron variant | No published information available. | |
| rs2781667 | Intron 1 | Intron variant | Major allele associated with higher severity of erectile dysfunction. | [68] |
| ARG2 | ||||
| rs3742879 | Intron 7 | Intron variant | Major allele associated with lower exhaled nitric oxide in children, and more severe airway obstruction in patients with asthma. | [69,70] |
| rs3759757 | 8 kb upstream variant | No published information available. | ||
| AGXT2 | ||||
| rs37369 | Exon 4 | Missense variant | Minor allele associated with higher methylarginine levels, higher diastolic blood pressure, and higher risk of coronary artery disease. | [71,72,73] |
| rs16899974 | Exon 14 | Missense variant | Minor allele associated with higher levels of SDMA, with atrial fibrillation and ischemic stroke, and higher risk of coronary artery disease. | [71,73,74] |
| PRMT1 | ||||
| rs10415880 | Intron 8 | Intron variant | Minor allele associated with arteriovenous fistula malfunction in male hemodialysis patients. | [75] |
| rs975484 | Upstream transcript variant | No published information available. |
Appendix B
| Gene/SNP | Major/Minor Allele | Allele Frequency Measured | Allele Frequency Expected # | p |
|---|---|---|---|---|
| NOS3 | ||||
| rs1799983 | G/T | 0.706/0.294 | 0.656/0.344 | 0.223 |
| rs2070744 | T/C | 0.598/0.402 | 0.562/0.438 | 0.530 |
| rs891512 | G/A | 0.843/0.157 | 0.751/0.249 | 0.039 |
| DDAH1 | ||||
| rs1241321 | A/G | 0.598/0.402 | 0.712/0.288 | 0.023 |
| rs233112 | T/C | 0.667/0.333 | 0.617/0.383 | 0.338 |
| rs480414 | G/A | 0.598/0.402 | 0.698/0.302 | 0.043 |
| DDAH2 | ||||
| rs805304 | T/G | 0.529/0.471 | 0.639/0.361 | 0.032 |
| rs2272592 | C/T | 0.892/0.108 | 0.844/0.156 | 0.246 |
| ARG1 | ||||
| rs2246012 | C/T | 0.853/0.147 | 0.680/0.320 | 0.677 |
| rs2781667 | T/C | 0.676/0.324 | 0.829/0.171 | 1.000 |
| ARG2 | ||||
| rs3742879 | G/C | 0.696/0.304 | 0.741/0.259 | 0.911 |
| rs3759757 | A/G | 0.755/0.245 | 0.687/0.313 | 0.813 |
| AGXT2 | ||||
| rs37369 | C/A | 0.075/0.925 | 0.773/0.227 | 1.000 |
| rs16899974 | C/T | 0.185/0.815 | 0.913/0.086 | 0.385 |
| PRMT1 | ||||
| rs10415880 | G/A | 0.407/0.593 | 0.738/0.262 | 0.158 |
| rs975484 | C/G | 0.685/0.315 | 0.658/0.342 | 0.197 |
References
- Harrod, C.G.; Bendok, B.R.; Batjer, H.H. Prediction of cerebral vasospasm in patients presenting with aneurysmal subarachnoid hemorrhage: A review. Neurosurgery 2005, 56, 633–654. [Google Scholar] [CrossRef]
- Miller, C.M.; Palestrant, D.; Schievink, W.I.; Alexander, M.J. Prolonged transcranial Doppler monitoring after aneurysmal subarachnoid hemorrhage fails to adequately predict ischemic risk. Neurocrit. Care 2011, 15, 387–392. [Google Scholar] [CrossRef]
- Bauknight, G.C., Jr.; Faraci, F.M.; Heistad, D.D. Endothelium-derived relaxing factor modulates noradrenergic constriction of cerebral arterioles in rabbits. Stroke 1992, 23, 1522–1525. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.M. Delayed cerebral vasospasm and nitric oxide: Review, new hypothesis, and proposed treatment. Pharmacol. Ther. 2005, 105, 23–56. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, Y.; Li, B.; Luo, C.; Zuo, S.; Liu, X.; Zhang, J.H.; Ruan, H.; Feng, H. Hemoglobin induced NO/cGMP suppression Deteriorate Microcirculation via Pericyte Phenotype Transformation after Subarachnoid Hemorrhage in Rats. Sci. Rep. 2016, 6, 22070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugelshofer, M.; Buzzi, R.M.; Schaer, C.A.; Richter, H.; Akeret, K.; Anagnostakou, V.; Mahmoudi, L.; Vaccani, R.; Vallelian, F.; Deuel, J.W.; et al. Haptoglobin administration into the subarachnoid space prevents hemoglobin-induced cerebral vasospasm. J. Clin. Investig. 2019, 129, 5219–5235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terpolilli, N.A.; Feiler, S.; Dienel, A.; Müller, F.; Heumos, N.; Friedrich, B.; Stover, J.; Schöller, K.; Plesnila, N. Nitric oxide inhalation reduces brain damage, prevents mortality, and improves neurological outcome after subarachnoid hemorrhage by resolving early pial microvasospasms. J. Cereb. Blood Flow Metab. 2016, 36, 2096–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradilla, G.; Garzon-Muvdi, T.; Ruzevick, J.J.; Bender, M.; Edwards, L.; Momin, E.N.; Thompson, R.C.; Tamargo, R.J. Systemic L-citrulline prevents cerebral vasospasm in haptoglobin 2-2 transgenic mice after subarachnoid hemorrhage. Neurosurgery 2012, 70, 747–756, discussion 756–757. [Google Scholar] [CrossRef]
- Böger, R.H.; Ron, E.S. L-Arginine improves vascular function by overcoming deleterious effects of ADMA, a novel cardiovascular risk factor. Altern. Med. Rev. 2005, 10, 14–23. [Google Scholar]
- Appel, D.; Seeberger, M.; Schwedhelm, E.; Czorlich, P.; Goetz, A.E.; Böger, R.H.; Hannemann, J. Asymmetric and symmetric dimethylarginines are markers of delayed cerebral ischemia and neurological outcome in patients with subarachnoid hemorrhage. Neurocrit. Care 2018, 29, 84–93. [Google Scholar] [CrossRef]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiper, J.M.; Santa Maria, J.; Chubb, A.; MacAllister, R.J.; Charles, I.G.; Whitley, G.S.; Vallance, P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem. J. 1999, 343 Pt 1, 209–214. [Google Scholar] [CrossRef]
- Lüneburg, N.; Lieb, W.; Zeller, T.; Chen, M.H.; Maas, R.; Carter, A.M.; Xanthakis, V.; Glazer, N.L.; Schwedhelm, E.; Sehsadri, S.; et al. Genome-wide association study of L-arginine and dimethylarginines reveals novel metabolic pathway for symmetric dimethylarginine. Circ. Cardiovasc. Genet. 2014, 7, 864–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Dimethylarginine:pyruvate aminotransferase in rats. Purification, properties, and identity with alanine: Glyoxylate aminotransferase 2. J. Biol. Chem. 1990, 265, 20938–20945. [Google Scholar] [PubMed]
- Böger, R.; Hannemann, J. Dual role of the L-arginine-ADMA-NO pathway in systemic hypoxic vasodilation and pulmonary hypoxic vasoconstriction. Pulm. Circ. 2020, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böger, R.H.; Maas, R.; Schulze, F.; Schwedhelm, E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality--An update on patient populations with a wide range of cardiovascular risk. Pharmacol. Res. 2009, 60, 481–487. [Google Scholar] [CrossRef]
- Böger, R.H.; Sullivan, L.M.; Schwedhelm, E.; Wang, T.J.; Maas, R.; Benjamin, E.J.; Schulze, F.; Xanthakis, V.; Benndorf, R.A.; Vasan, R.S. Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation 2009, 119, 1592–1600. [Google Scholar] [CrossRef]
- Lüneburg, N.; von Holten, R.A.; Töpper, R.F.; Schwedhelm, E.; Maas, R.; Böger, R.H. Symmetric dimethylarginine is a marker of detrimental outcome in the acute phase after ischaemic stroke: Role of renal function. Clin. Sci. 2012, 122, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Schulze, F.; Carter, A.M.; Schwedhelm, E.; Ajjan, R.; Maas, R.; von Holten, R.A.; Atzler, D.; Grant, P.J.; Böger, R.H. Symmetric dimethylarginine predicts all-cause mortality following ischemic stroke. Atherosclerosis 2010, 208, 518–523. [Google Scholar] [CrossRef]
- Jung, C.S.; Oldfield, E.H.; Harvey-White, J.; Espey, M.G.; Zimmermann, M.; Seifert, V.; Pluta, R.M. Association of an endogenous inhibitor of nitric oxide synthase with cerebral vasospasm in patients with aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2007, 107, 945–950. [Google Scholar] [CrossRef]
- Li, H.; Wu, W.; Liu, M.; Zhang, X.; Zhang, Q.R.; Ni, L.; Hang, C.H. Increased cerebrospinal fluid concentrations of asymmetric dimethylarginine correlate with adverse clinical outcome in subarachnoid hemorrhage patients. J. Clin. Neurosci. 2014, 21, 1404–1408. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, C.; Hultin, M.; Koskinen, L.O.; Lindvall, P.; Borota, L.; Naredi, S. ADMA levels and arginine/ADMA ratios reflect severity of disease and extent of inflammation after subarachnoid hemorrhage. Neurocrit. Care 2014, 21, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.S.; Iuliano, B.A.; Harvey-White, J.; Espey, M.G.; Oldfield, E.H.; Pluta, R.M. Association between cerebrospinal fluid levels of asymmetric dimethyl-L-arginine, an endogenous inhibitor of endothelial nitric oxide synthase, and cerebral vasospasm in a primate model of subarachnoid hemorrhage. J. Neurosurg. 2004, 101, 836–842. [Google Scholar] [CrossRef] [Green Version]
- Ko, N.U.; Rajendran, P.; Kim, H.; Rutkowski, M.; Pawlikowska, L.; Kwok, P.Y.; Higashida, R.T.; Lawton, M.T.; Smith, W.S.; Zaroff, J.G.; et al. Endothelial nitric oxide synthase polymorphism (-786T->C) and increased risk of angiographic vasospasm after aneurysmal subarachnoid hemorrhage. Stroke 2008, 39, 1103–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starke, R.M.; Kim, G.H.; Komotar, R.J.; Hickman, Z.L.; Black, E.M.; Rosales, M.B.; Kellner, C.P.; Hahn, D.K.; Otten, M.L.; Edwards, J.; et al. Endothelial nitric oxide synthase gene single-nucleotide polymorphism predicts cerebral vasospasm after aneurysmal subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2008, 28, 1204–1211. [Google Scholar] [CrossRef]
- Alexander, S.; Poloyac, S.; Hoffman, L.; Gallek, M.; Dianxu, R.; Balzer, J.; Kassam, A.; Conley, Y. Endothelial nitric oxide synthase tagging single nucleotide polymorphisms and recovery from aneurysmal subarachnoid hemorrhage. Biol. Res. Nurs. 2009, 11, 42–52. [Google Scholar] [CrossRef]
- Abhary, S.; Burdon, K.P.; Kuot, A.; Javadiyan, S.; Whiting, M.J.; Kasmeridis, N.; Petrovsky, N.; Craig, J.E. Sequence variation in DDAH1 and DDAH2 genes is strongly and additively associated with serum ADMA concentrations in individuals with type 2 diabetes. PLoS ONE 2010, 5, e9462. [Google Scholar] [CrossRef] [Green Version]
- Caplin, B.; Nitsch, D.; Gill, H.; Hoefield, R.; Blackwell, S.; MacKenzie, D.; Cooper, J.A.; Middleton, R.J.; Talmud, P.J.; Veitch, P.; et al. Circulating methylarginine levels and the decline in renal function in patients with chronic kidney disease are modulated by DDAH1 polymorphisms. Kidney Int. 2010, 77, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Thaha, M.; Nilamsari, W.P.; Yusuf, M.; Amin, M. Profile of PRMT-1 gene polymorphism in hemodialysis patients with increased ADMA levels. Acta Med. Indones. 2014, 46, 97–103. [Google Scholar]
- Hannemann, J.; Zummack, J.; Hillig, J.; Böger, R. Metabolism of asymmetric dimethylarginine in hypoxia: From bench to bedside. Pulm. Circ. 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Connolly, E.S., Jr.; Rabinstein, A.A.; Carhuapoma, J.R.; Derdeyn, C.P.; Dion, J.; Higashida, R.T.; Hoh, B.L.; KIrkness, C.J.; Naidech, A.M.; Ogilvy, C.S.; et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012, 43, 1711–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diringer, M.N.; Bleck, T.P.; Claude Hemphill, J., 3rd; Menon, D.; Shutter, L.; Vespa, P.; Bruder, N.; Connolly, E.S., Jr.; Citerio, G.; Gress, D.; et al. Critical care management of patients following aneurysmal subarachnoid hemorrhage: Recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit. Care 2011, 15, 211–240. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.G.; Hunt, W.E.; Sano, K. Report of World Federation of Neurological Surgeons Committee on a Universal Subarachnoid Hemorrhage Grading Scale. J. Neurosurg. 1988, 68, 985–986. [Google Scholar] [CrossRef]
- Gotoh, O.; Tamura, A.; Yasui, N.; Suzuki, A.; Hadeishi, H.; Sano, K. Glasgow Coma Scale in the prediction of outcome after early aneurysm surgery. Neurosurgery 1996, 39, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Hunt, W.E.; Hess, R.M. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J. Neurosurg. 1968, 28, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, C.M.; Kistler, J.P.; Davis, J.M. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980, 6, 1–9. [Google Scholar] [CrossRef]
- Vergouwen, M.D.; Vermeulen, M.; van Gijn, J.; Rinkel, G.J.; Wijdicks, E.F.; Muizelaar, J.P.; Mendelow, A.D.; Juvela, S.; Yonas, H.; Terbrugge, K.G.; et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: Proposal of a multidisciplinary research group. Stroke 2010, 41, 2391–2395. [Google Scholar] [CrossRef] [Green Version]
- Westermaier, T.; Pham, M.; Stetter, C.; Willner, N.; Solymosi, L.; Ernestus, R.I.; Vince, G.H.; Kunze, E. Value of transcranial Doppler, perfusion-CT and neurological evaluation to forecast secondary ischemia after aneurysmal SAH. Neurocrit. Care 2014, 20, 406–412. [Google Scholar] [CrossRef]
- Machiela, M.J.; Chanock, S.J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef]
- Schwedhelm, E.; Tan-Andresen, J.; Maas, R.; Riederer, U.; Schulze, F.; Böger, R.H. Liquid chromatography-tandem mass spectrometry method for the analysis of asymmetric dimethylarginine in human plasma. Clin. Chem. 2005, 51, 1268–1271. [Google Scholar] [CrossRef] [Green Version]
- Böger, R.H.; Sydow, K.; Borlak, J.; Thum, T.; Lenzen, H.; Schubert, B.; Tsikas, D.; Bode-Böger, S.M. LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: Involvement of S-adenosylmethionine-dependent methyltransferases. Circ. Res. 2000, 87, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pikula, A.; Böger, R.H.; Beiser, A.S.; Maas, R.; DeCarli, C.; Schwedhelm, E.; Himali, J.J.; Schulze, F.; Au, R. Kelly-Hayes, M.; et al. Association of plasma ADMA levels with MRI markers of vascular brain injury: Framingham offspring study. Stroke 2009, 40, 2959–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodionov, R.N.; Murry, D.J.; Vaulman, S.F.; Stevens, J.W.; Lentz, S.R. Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethylarginine and protects from inhibition of nitric oxide production. J. Biol. Chem. 2010, 285, 5385–5391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlt, S.; Schulze, F.; Eichenlaub, M.; Maas, R.; Lehmbeck, J.T.; Schwedhelm, E.; Jahn, H.; Böger, R.H. Asymmetrical dimethylarginine is increased in plasma and decreased in cerebrospinal fluid of patients with Alzheimer’s disease. Dement Geriatr. Cogn. Disord. 2008, 26, 58–64. [Google Scholar] [CrossRef]
- Xu, L.; Wang, B.; Kaur, K.; Kho, M.F.; Cooke, J.P.; Giffard, R.G. NOx and ADMA changes with focal ischemia, amelioration with the chaperonin GroEL. Neurosci. Lett. 2007, 418, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Brouns, R.; Marescau, B.; Possemiers, I.; Sheorajpanday, R.; De Deyn, P.P. Dimethylarginine levels in cerebrospinal fluid of hyperacute ischemic stroke patients are associated with stroke severity. Neurochem. Res. 2009, 34, 1642–1649. [Google Scholar] [CrossRef]
- Closs, E.I.; Basha, F.Z.; Habermeier, A.; Förstermann, U. Interference of L-arginine analogues with L-arginine transport mediated by the y+ carrier hCAT-2B. Nitric Oxide 1997, 1, 65–73. [Google Scholar] [CrossRef]
- McBride, A.E.; Silver, P.A. State of the arg: Protein methylation at arginine comes of age. Cell 2001, 106, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Malik, R.; Rannikmae, K.; Traylor, M.; Georgakis, M.K.; Sargurupremraj, M.; Markus, H.S.; Hopewell, J.C.; Debette, S.; Sudlow, C.L.M.; Dichgans, M. Genome-wide meta-analysis identifies 3 novel loci associated with stroke. Ann. Neurol. 2018, 84, 934–939. [Google Scholar] [CrossRef]
- Morris, C.M.; Ballard, C.G.; Allan, L.; Rowan, E.; Stephens, S.; Firbank, M.; Ford, G.A.; Kenny, R.A.; O’Brien, J.T.; Kalaria, R.N. NOS3 gene rs1799983 polymorphism and incident dementia in elderly stroke survivors. Neurobiol. Aging 2011, 32, e551–e556. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, X.; Wu, W.; Zhang, D. Association of G894T polymorphism in endothelial nitric oxide synthase gene with the risk of ischemic stroke: A meta-analysis. Biomed. Rep. 2013, 1, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lynch, A.I.; Davis, B.R.; Ford, C.E.; Boerwinkle, E.; Eckfeldt, J.H.; Leiendecker-Foster, C.; Arnett, D.K. Pharmacogenetic association of NOS3 variants with cardiovascular disease in patients with hypertension: The GENHAT study. PLoS ONE 2012, 7, e34217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrix, P.; Foreman, P.M.; Harrigan, M.R.; Fisher, W.S., 3rd; Vyas, N.A.; Lipsky, R.H.; Lin, M.; Walters, B.C.; Tubbs, R.S.; Shoja, M.M.; et al. Endothelial nitric oxide synthase polymorphism is associated with delayed cerebral ischemia following aneurysmal subarachnoid hemorrhage. World Neurosurg. 2017, 101, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, P.; Foreman, P.M.; Harrigan, M.R.; Fisher, W.S.R.; Vyas, N.A.; Lipsky, R.H.; Lin, M.; Walters, B.C.; Tubbs, R.S.; Shoja, M.M.; et al. The role of endothelial nitric oxide synthase -786 T/C polymorphism in cardiac instability following aneurysmal subarachnoid hemorrhage. Nitric Oxide 2017, 71, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kato, K.; Oguri, M.; Yoshida, T.; Yokoi, K.; Watanabe, S.; Metoki, N.; Yoshida, H.; Satoh, K.; Ichihara, S.; et al. Association of genetic variants with atherothrombotic cerebral infarction in Japanese individuals with metabolic syndrome. Int. J. Mol. Med. 2008, 21, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Kardia, S.L.; Greene, M.T.; Boerwinkle, E.; Turner, S.T.; Kullo, I.J. Investigating the complex genetic architecture of ankle-brachial index, a measure of peripheral arterial disease, in non-hispanic whites. BMC Med. Genom. 2008, 1, 16. [Google Scholar] [CrossRef] [Green Version]
- Kullo, I.J.; Greene, M.T.; Boerwinkle, E.; Chu, J.; Turner, S.T.; Kardia, S.L. Association of polymorphisms in NOS3 with the ankle-brachial index in hypertensive adults. Atherosclerosis 2008, 196, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lin, Y.; Zhang, R. Associations between endothelial nitric oxide synthase gene polymorphisms and the risk of coronary artery disease: A systematic review and meta-analysis of 132 case-control studies. Eur. J. Prev. Cardiol. 2019, 26, 160–170. [Google Scholar] [CrossRef]
- Seelenfreund, D.; Lobos, S.; Quesada, A.; Saavedra, J.; Wolff, C.; López-Stewart, G.; Araya, A.; Durruty, P. Association of the intronic polymorphism rs891512 (G24943A) of the endothelial nitric oxide synthase gene with hypertension in Chilean type 2 diabetes patients. Diabetes Res. Clin. Pract. 2012, 96, e47–e49. [Google Scholar] [CrossRef]
- Vimaleswaran, K.S.; Franks, P.W.; Barroso, I.; Brage, S.; Ekelund, U.; Wareham, N.J.; Loos, R.J.F. Habitual energy expenditure modifies the association between NOS3 gene polymorphisms and blood pressure. Am. J. Hypertens 2008, 21, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.M.; Lin, S.J.; Lin, M.W.; Hsu, C.P.; Chung, M.Y. The association of dimethylarginine dimethylaminohydrolase 1 gene polymorphism with type 2 diabetes: A cohort study. Cardiovasc. Diabetol. 2011, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnabel, R.; Larson, M.G.; Dupuis, J.; Lunetta, K.L.; Lipinska, I.; Meigs, J.B.; Yin, X.; Rong, Y.; Vita, J.A.; Newton-Cheh, C.; et al. Relations of inflammatory biomarkers and common genetic variants with arterial stiffness and wave reflection. Hypertension 2008, 51, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Trittmann, J.K.; Gastier-Foster, J.M.; Zmuda, E.J.; Frick, J.; Rogers, L.K.; Vieland, V.J.; Chicoine, L.G.; Nelin, L.D. A single nucleotide polymorphism in the dimethylarginine dimethylaminohydrolase gene is associated with lower risk of pulmonary hypertension in bronchopulmonary dysplasia. Acta Paediatr. 2016, 105, e170–e175. [Google Scholar] [CrossRef] [Green Version]
- Maas, R.; Erdmann, J.; Lüneburg, N.; Stritzke, J.; Schwedhelm, E.; Meisinger, C.; Peters, A.; Weil, J.; Schunkert, H.; Böger, R.H.; et al. Polymorphisms in the promoter region of the dimethylarginine dimethylaminohydrolase 2 gene are associated with prevalence of hypertension. Pharmacol Res. 2009, 60, 488–493. [Google Scholar] [CrossRef]
- Marra, M.; Marchegiani, F.; Ceriello, A.; Sirolla, C.; Boemi, M.; Franceschi, C.; Spazzafumo, L.; Testa, I.; Bonfigli, A.R.; Cucchi, M.; et al. Chronic renal impairment and DDAH2-1151 a/c polymorphism determine ADMA levels in type 2 diabetic subjects. Nephrol. Dial. Transpl. 2013, 28, 964–971. [Google Scholar] [CrossRef] [Green Version]
- Perez-Hernandez, N.; Vargas-Alarcon, G.; Arellano-Zapoteco, R.; Martinez-Rodriguez, N.; Fragoso, J.M.; Aptilon-Duque, G.; Posadas-Sanchez, R.; Posadas-Romero, C.; Juarez-Cedillo, T.; Dominguez-Lopez, M.L.; et al. Protective role of DDAH2 (rs805304) gene polymorphism in patients with myocardial infarction. Exp. Mol. Pathol. 2014, 97, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.A.; Kim, S.W.; Jeon, E.J.; Jeong, J.Y.; Moon, S.S.; Lee, W.K.; Kim, J.G.; Lee, I.K.; Park, K.G. Association of the DDAH2 gene polymorphism with type 2 diabetes and hypertension. Diabetes Res. Clin. Pract. 2012, 98, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Lacchini, R.; Muniz, J.J.; Nobre, Y.T.; Cologna, A.J.; Martins, A.C.; Tanus-Santos, J.E. Relationship between arginase 1 and arginase 2 levels and genetic polymorphisms with erectile dysfunction. Nitric Oxide 2015, 51, 36–42. [Google Scholar] [CrossRef]
- Salam, M.T.; Bastain, T.M.; Rappaport, E.B.; Islam, T.; Berhane, K.; Gauderman, W.J.; Gilliland, F.D. Genetic variations in nitric oxide synthase and arginase influence exhaled nitric oxide levels in children. Allergy 2011, 66, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Vonk, J.M.; Postma, D.S.; Maarsingh, H.; Bruinenberg, M.; Koppelman, G.H.; Meurs, H. Arginase 1 and arginase 2 variations associate with asthma, asthma severity and beta2 agonist and steroid response. Pharm. Genom. 2010, 20, 179–186. [Google Scholar] [CrossRef]
- Amir, M.; Hassanein, S.I.; Abdel Rahman, M.F.; Gad, M.Z. AGXT2 and DDAH-1 genetic variants are highly correlated with serum ADMA and SDMA levels and with incidence of coronary artery disease in Egyptians. Mol. Biol. Rep. 2018, 45, 2411–2419. [Google Scholar] [CrossRef] [PubMed]
- Caplin, B.; Wang, Z.; Slaviero, A.; Tomlinson, J.; Dowsett, L.; Delahaye, M.; Salama, A.; Wheeler, D.C.; Leiper, J. Alanine-glyoxylate aminotransferase-2 metabolizes endogenous methylarginines, regulates NO, and controls blood pressure. Arter. Thromb. Vasc. Biol. 2012, 32, 2892–2900. [Google Scholar] [CrossRef] [Green Version]
- Kittel, A.; Müller, F.; König, J.; Mieth, M.; Sticht, H.; Zolk, O.; Kralj, A.; Heinrich, M.R.; Fromm, M.F.; Maas, R. Alanine-glyoxylate aminotransferase 2 (AGXT2) polymorphisms have considerable impact on methylarginine and beta-aminoisobutyrate metabolism in healthy volunteers. PLoS ONE 2014, 9, e88544. [Google Scholar] [CrossRef] [PubMed]
- Seppälä, I.; Kleber, M.E.; Bevan, S.; Lyytikäinen, L.P.; Oksala, N.; Hernesniemi, J.A.; Mäkelä, K.M.; Rothwell, P.M.; Sudlow, C.; Dichgans, M.; et al. Associations of functional alanine-glyoxylate aminotransferase 2 gene variants with atrial fibrillation and ischemic stroke. Sci. Rep. 2016, 6, 23207. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Tsai, W.J.; Chen, Y.W.; Yang, W.C.; Lee, C.Y.; Ou, S.M.; Chen, Y.T.; Chien, C.C.; Lee, P.C.; Chung, M.Y.; et al. Genotype polymorphisms of genes regulating nitric oxide synthesis determine long-term arteriovenous fistula patency in male hemodialysis patients. Ren. Fail. 2016, 38, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]




| All | Patients with DCI | Patients without DCI | p | |
|---|---|---|---|---|
| Demographics | ||||
| Number of patients | 51 | 18 | 33 | |
| Age, years (mean ± SD) | 54.8 ± 13.1 | 58.6 ± 14.1 | 52.6 ± 12.2 | 0.140 |
| Sex, male/female (%) | 13/38 (25.5/75.5) | 2/16 (11.2/88.8) | 11/22 (33.3/66.7) | 0.102 |
| BMI, kg/m2 (mean ± SD) | 25.9 ± 3.9 | 26.8 ± 4.9 | 25.4 ± 3.2 | 0.333 |
| Risk factors, n (%) | ||||
| Hypertension | 21 (41.2) | 9 (50.0) | 12 (36.4) | 0.385 |
| Diabetes mellitus | 3 (5.9) | 1 (5.6) | 2 (6.1) | 1.000 |
| Renal failure | 1 (2.0) | 1 (5.6) | 0 (0.0) | 0.353 |
| Smoker, current + ex | 11 (21.6) | 4 (22.2) | 7 (21.2) | 1.000 |
| Long-term medication, n (%) | ||||
| Angiotensin II blockers | 6 (11.8) | 2 (11.2) | 4 (12.1) | 0.586 |
| Beta blockers | 8 (15.7) | 3 (16.7) | 5 (15.2) | 0.591 |
| Diuretics | 3 (5.9) | 1 (5.6) | 2 (6.1) | 0.718 |
| Calcium channel blockers | 5 (9.8) | 2 (11.2) | 3 (9.1) | 0.585 |
| Statins | 6 (11.8) | 2 (11.2) | 4 (12.1) | 0.646 |
| NSAR | 3 (5.9) | 1 (5.6) | 2 (6.1) | 0.718 |
| Platelet inhibitors | 5 (9.8) | 2 (11.2) | 3 (9.1) | 0.585 |
| Oral anticoagulants | 1 (2.0) | 0 (0.0) | 1 (3.0) | 0.647 |
| Bleeding location, n (%) | ||||
| Anterior cerebral circulation | 35 (64.8) | 12 (66.7) | 23 (69.7) | 0.532 |
| Posterior cerebral circulation | 12 (22.2) | 4 (22.2) | 8 (24.2) | 0.579 |
| Aneurysm treatment, n (%) | ||||
| Clipping | 15 (29.4) | 6 (33.3) | 9 (27.3) | 0.442 |
| Coiling | 28 (54.9) | 9 (50.0) | 18 (54.5) | 0.492 |
| Non-interventional | 8 (15.7) | 2 (11.1) | 6 (18.2) | 0.409 |
| Nimodipin treatment | 47 (92.2) | 17 (94.4) | 30 (90.9) | 0.557 |
| All | Patients with DCI | Patients without DCI | p | |
|---|---|---|---|---|
| Number of patients | 51 | 18 | 33 | |
| Scores at admission (mean ± SD) | ||||
| GCS | 8.6 ± 5.6 | 10.9 ± 4.6 | 7.2 ± 5.7 | 0.021 |
| Hunt and Hess | 3.1 ± 1.4 | 2.9 ± 1.2 | 3.2 ± 1.5 | 0.622 |
| Fisher Score | 3.3 ± 0.7 | 3.2 ± 0.8 | 3.4 ± 0.7 | 0.416 |
| WFNS | 3.4 ± 1.7 | 2.9 ± 1.5 | 3.6 ± 1.8 | 0.134 |
| Outcome, n (%) | ||||
| Vasospasm | 22 (43.1) | 10 (55.6) | 12 (36.4) | 0.242 |
| In-hospital lethality | 7 (13.7) | 3 (16.7) | 4 (12.1) | 0.686 |
| Duration of ICU treatment (d) | 21.0 ± 11.5 | 22.7 ± 11.2 | 20.1 ± 11.7 | 0.449 |
| Mechanical ventilation (h) | 237.5 ± 272.5 | 313.3 ± 251.7 | 196.1 ± 278.2 | 0.144 |
| Re-intervention rate | 5 (9.8) | 4 (22.2) | 1 (3.0) | 0.047 |
| GOS at discharge | 3.4 ± 1.6 | 2.8 ± 1.6 | 3.6 ± 1.5 | 0.056 |
| GOS three months later | 3.6 ± 1.1 | 3.1 ± 1.1 | 4.0 ± 1.0 | 0.071 |
| DDAH1 Haplotype | Relative Risk | 95% CI | p |
|---|---|---|---|
| (3× homozygous major versus 0× homozygous minor) | |||
| DCI | 1.103 | 0.925–1.315 | 0.273 |
| Vasospasm | 1.483 | 1.013–2.170 | 0.039 |
| Death | 1.000 | 0.898–1.113 | 1.000 |
| GOS < 4 at discharge | 1.364 | 1.105–1.683 | 0.003 |
| GOS < 4 after 3 months | 1.475 | 1.231–1.766 | <0.0001 |
| (3× homozygous major versus ≥ 1× homozygous minor) | |||
| DCI | 1.316 | 1.072–1.614 | 0.011 |
| Vasospasm | 0.983 | 0.774–1.247 | 1.000 |
| Death | 0.929 | 0.460–1.874 | 1.000 |
| GOS < 4 at discharge | 2.273 | 1.681–3.072 | <0.0001 |
| GOS < 4 after 3 months | 1.299 | 1.110–1.519 | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hannemann, J.; Appel, D.; Seeberger-Steinmeister, M.; Brüning, T.; Zummack, J.; Böger, R. Sequence Variation in the DDAH1 Gene Predisposes for Delayed Cerebral Ischemia in Subarachnoidal Hemorrhage. J. Clin. Med. 2020, 9, 3900. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9123900
Hannemann J, Appel D, Seeberger-Steinmeister M, Brüning T, Zummack J, Böger R. Sequence Variation in the DDAH1 Gene Predisposes for Delayed Cerebral Ischemia in Subarachnoidal Hemorrhage. Journal of Clinical Medicine. 2020; 9(12):3900. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9123900
Chicago/Turabian StyleHannemann, Juliane, Daniel Appel, Miriam Seeberger-Steinmeister, Tabea Brüning, Julia Zummack, and Rainer Böger. 2020. "Sequence Variation in the DDAH1 Gene Predisposes for Delayed Cerebral Ischemia in Subarachnoidal Hemorrhage" Journal of Clinical Medicine 9, no. 12: 3900. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9123900