Oral Microbiome Signatures in Hematological Cancers Reveal Predominance of Actinomyces and Rothia Species

Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Sample Collection, Bacterial DNA Extraction and Sequencing
2.3. Data Preprocessing
2.4. Statistical Analysis
2.4.1. PERMANOVA
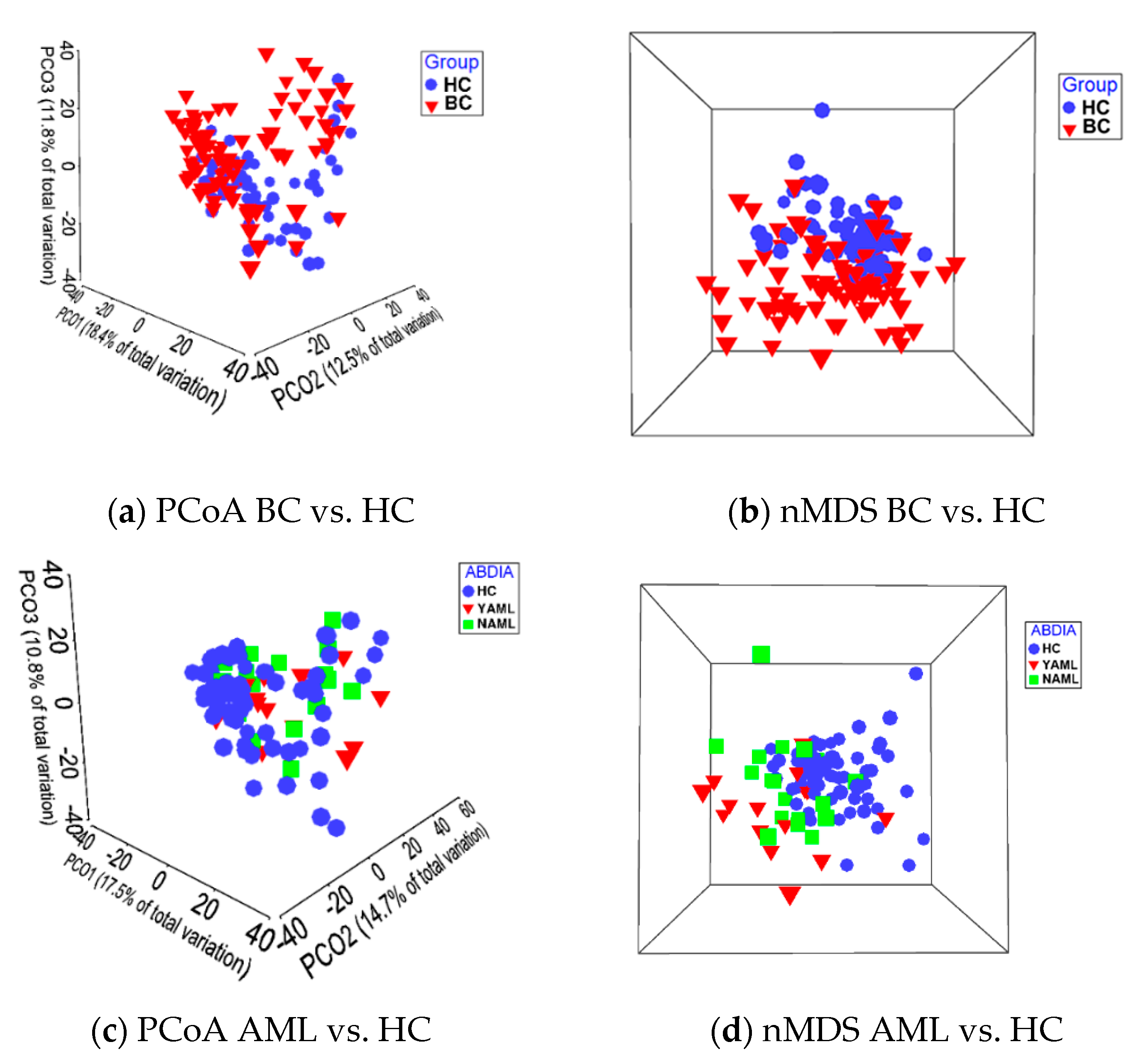
2.4.2. Principal Coordinate Analysis (PCoA) and Nonmetric Multidimensional Scaling (nMDS)
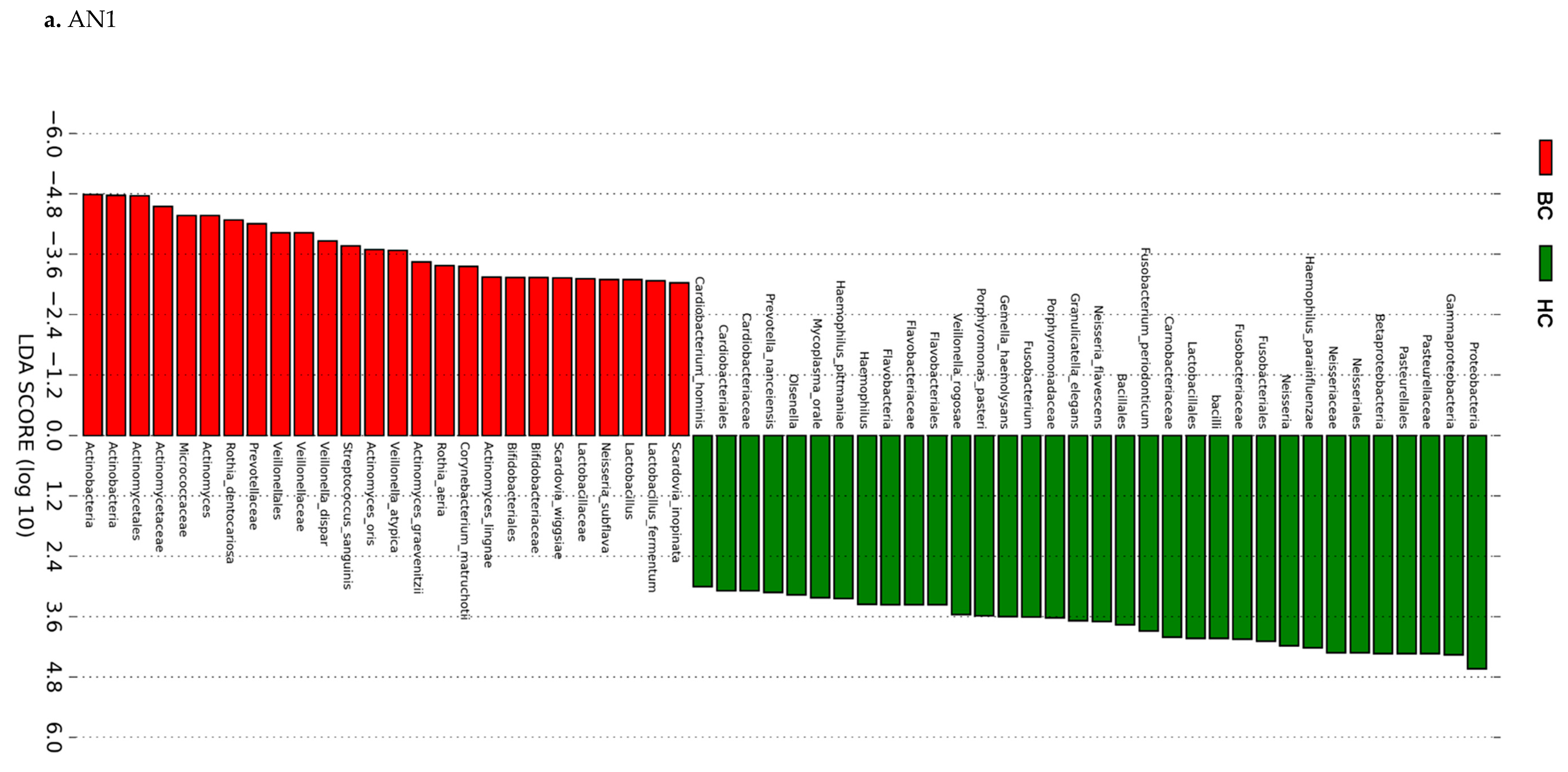
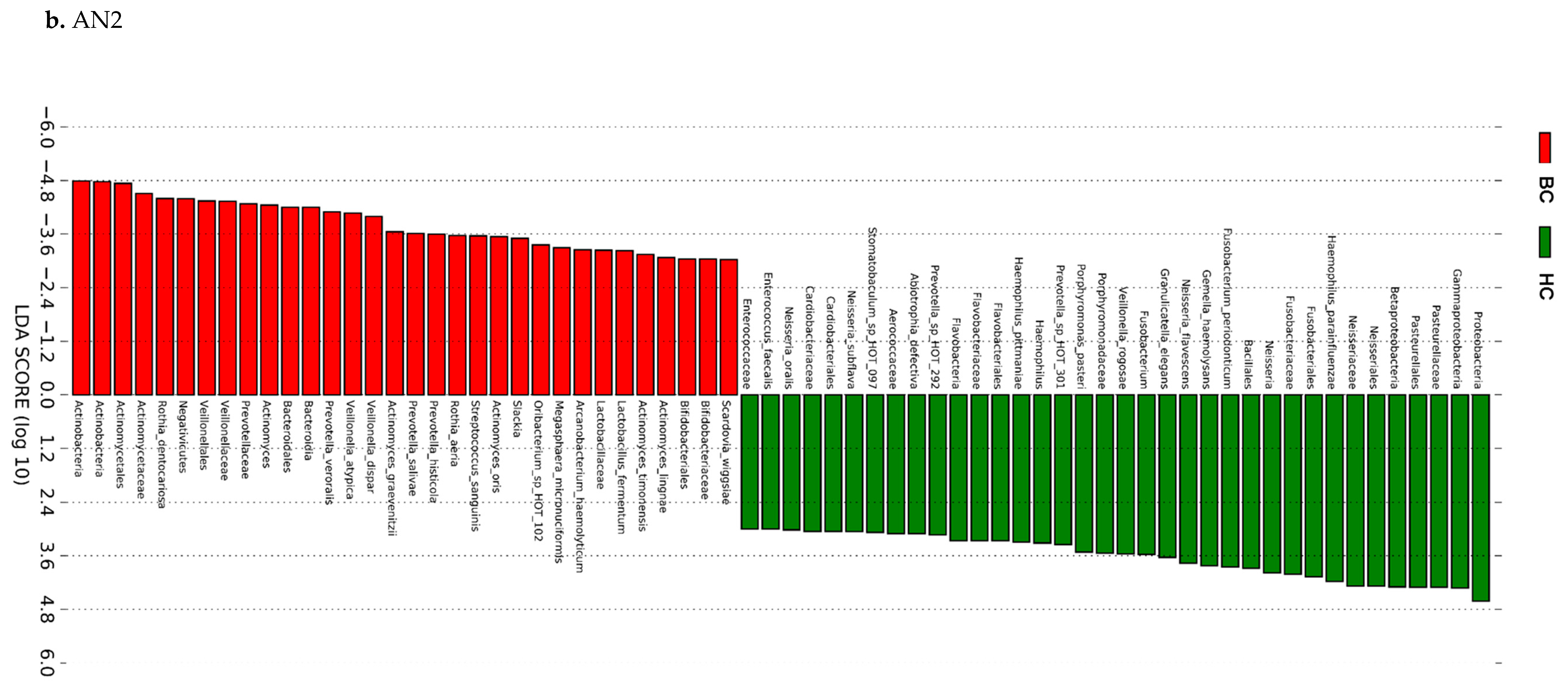
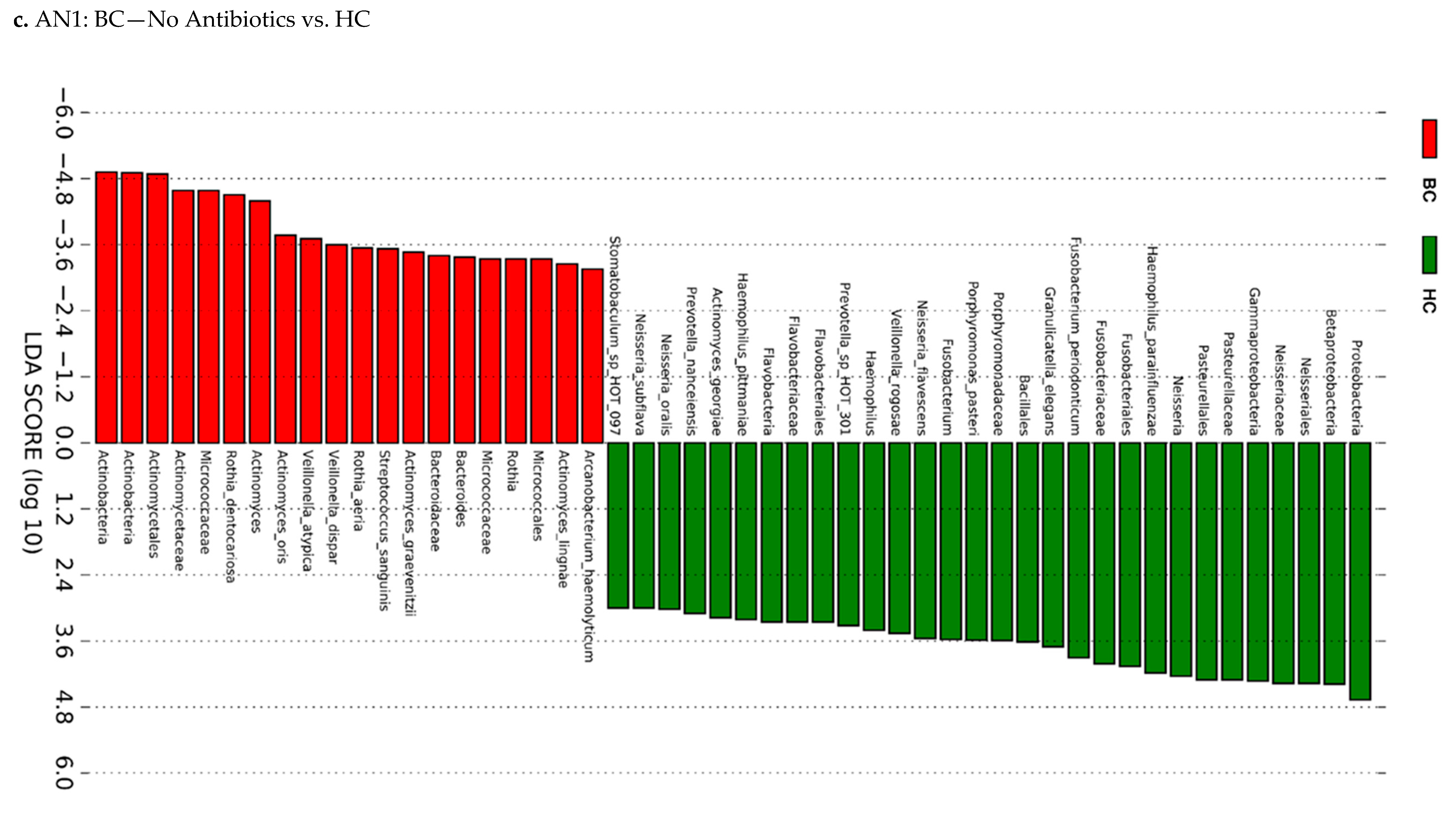
2.4.3. Linear Discriminant Analysis (LDA) Effect Size (LEfSe)
2.4.4. Alpha-Diversity
2.4.5. Association between Taxonomic Profiles and Pathogenicity
2.5. Ethics
3. Results
3.1. BC pts Were Found to Have a Reduction in Alpha-Diversity When Compared to HC
3.2. Beta-Diversity Shows Significant Difference between the BC and HC Groups
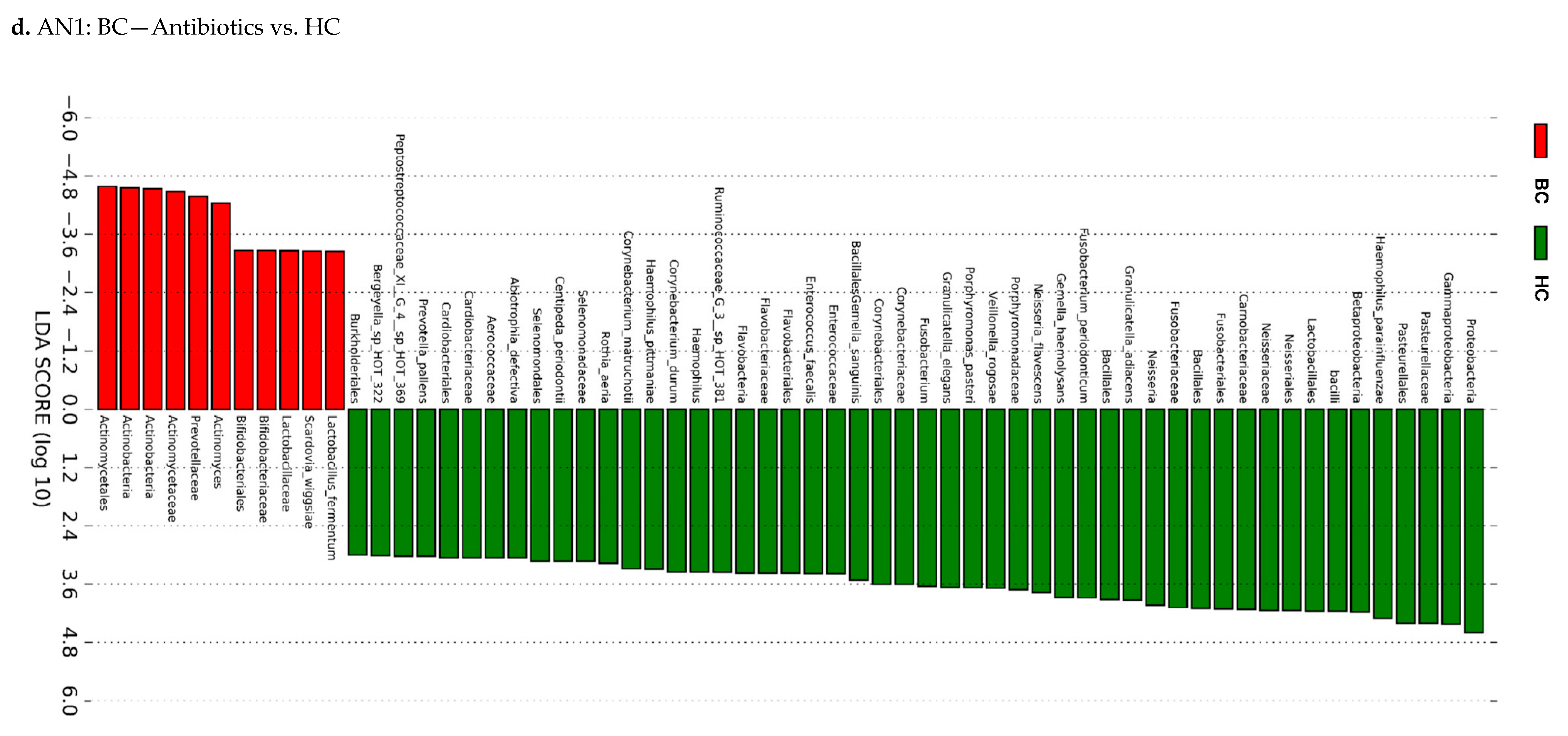
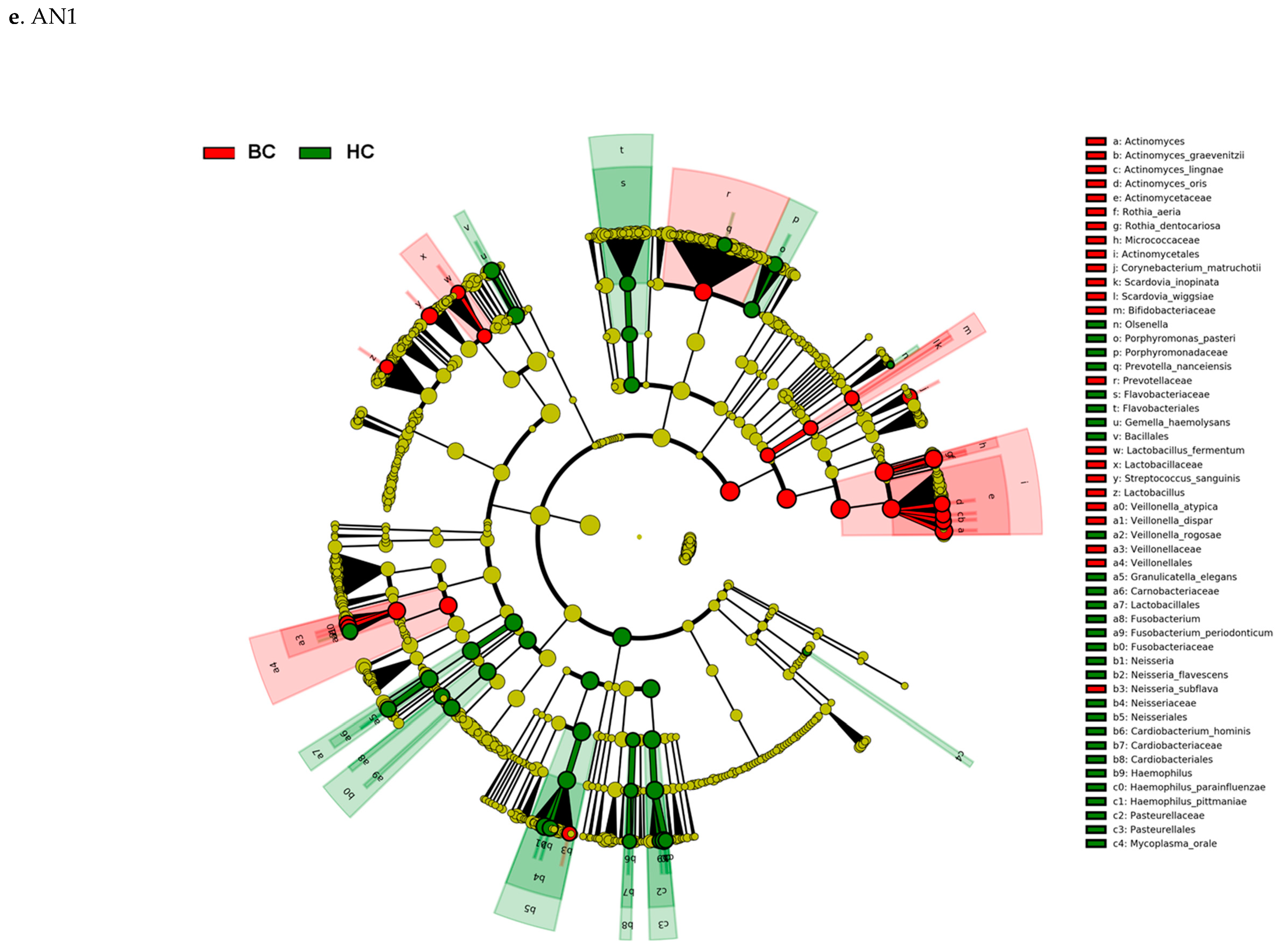
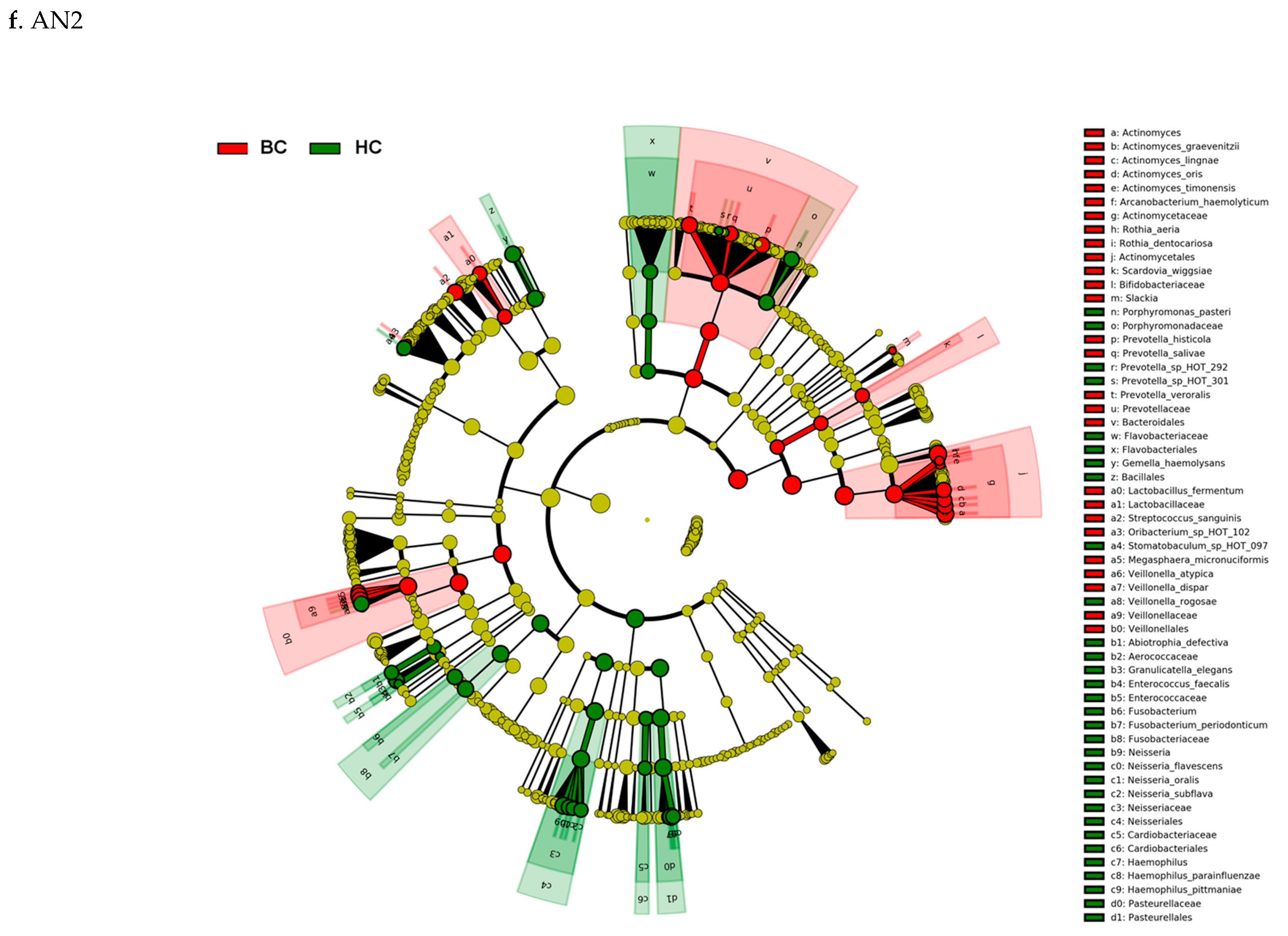
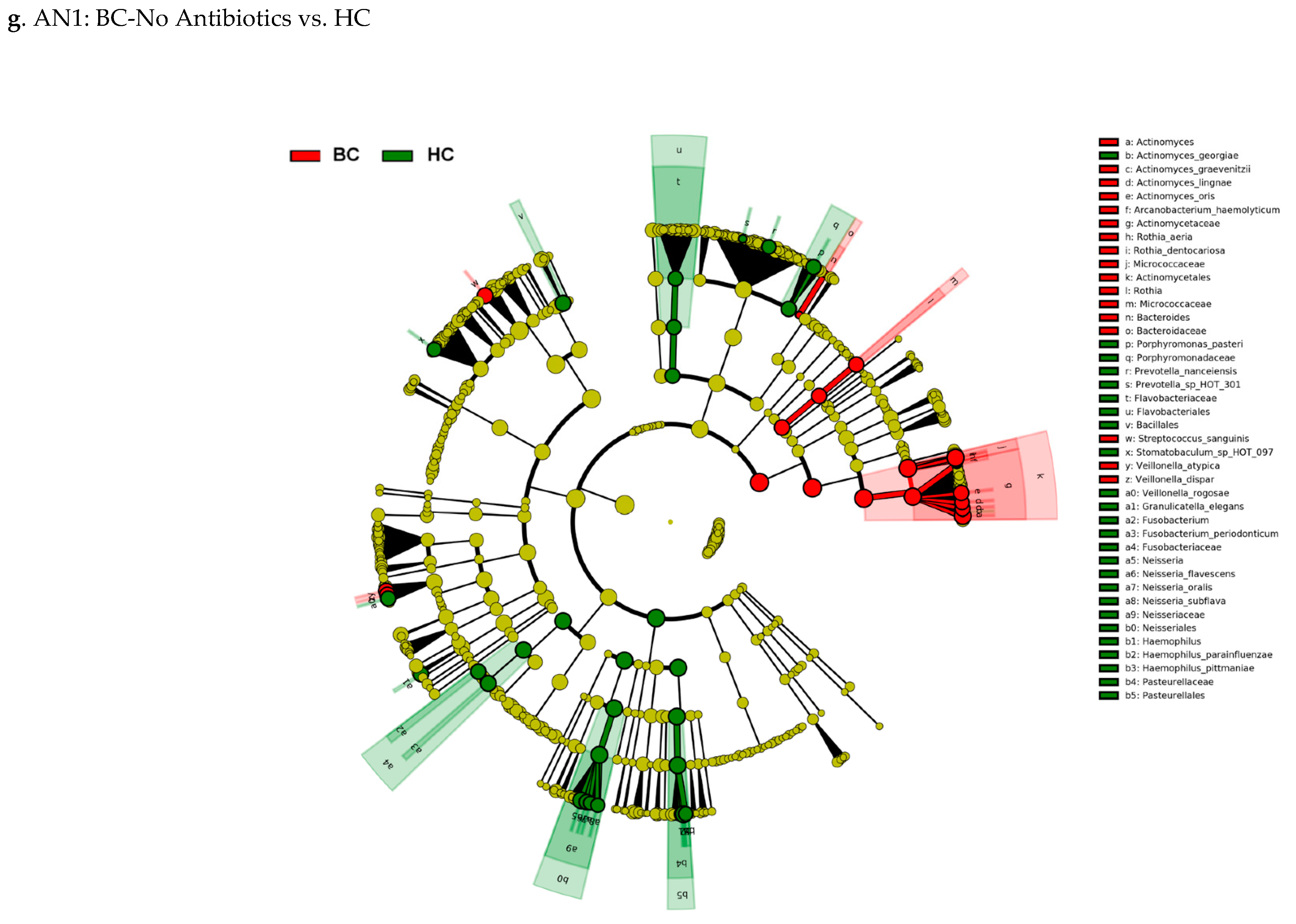
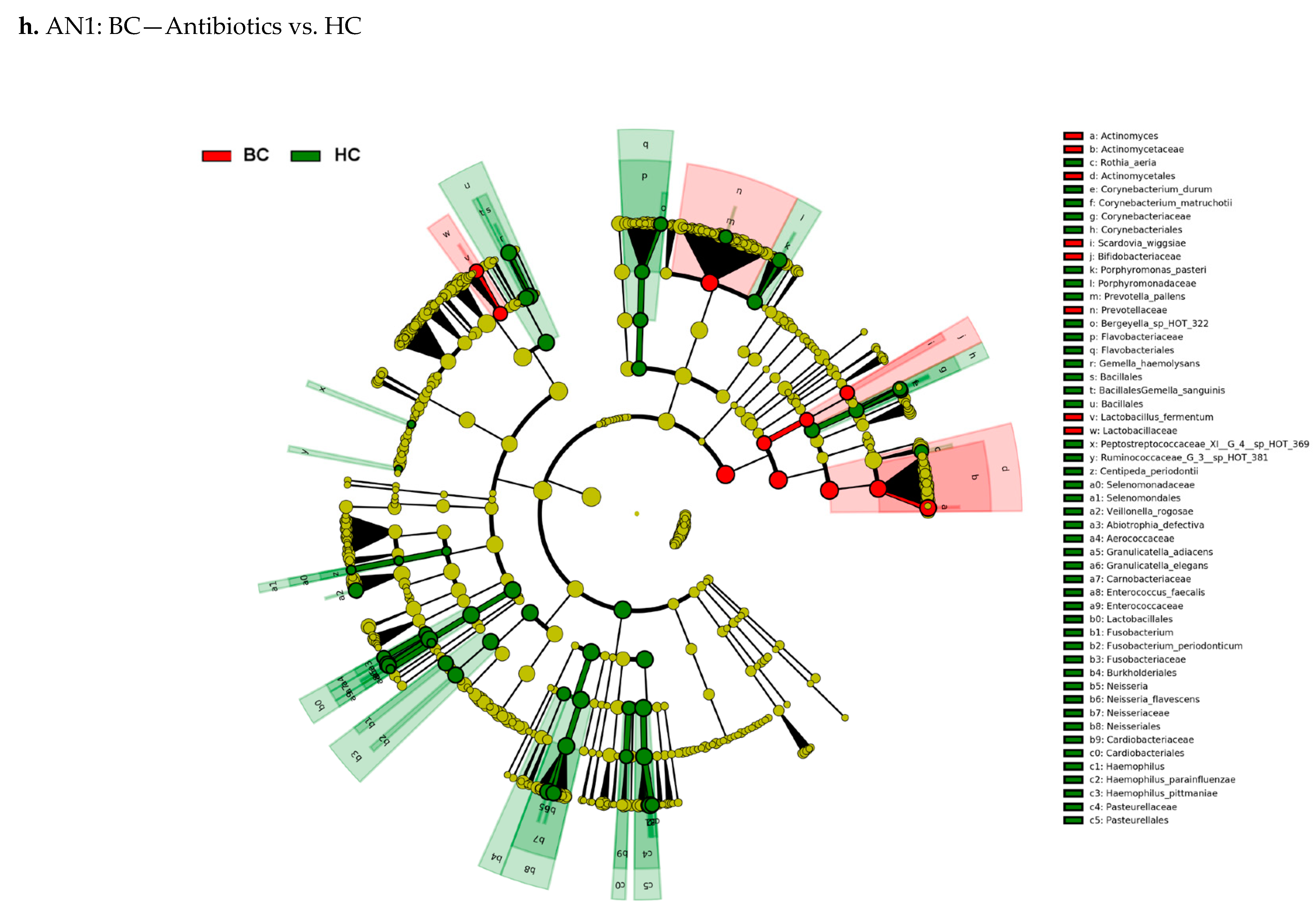
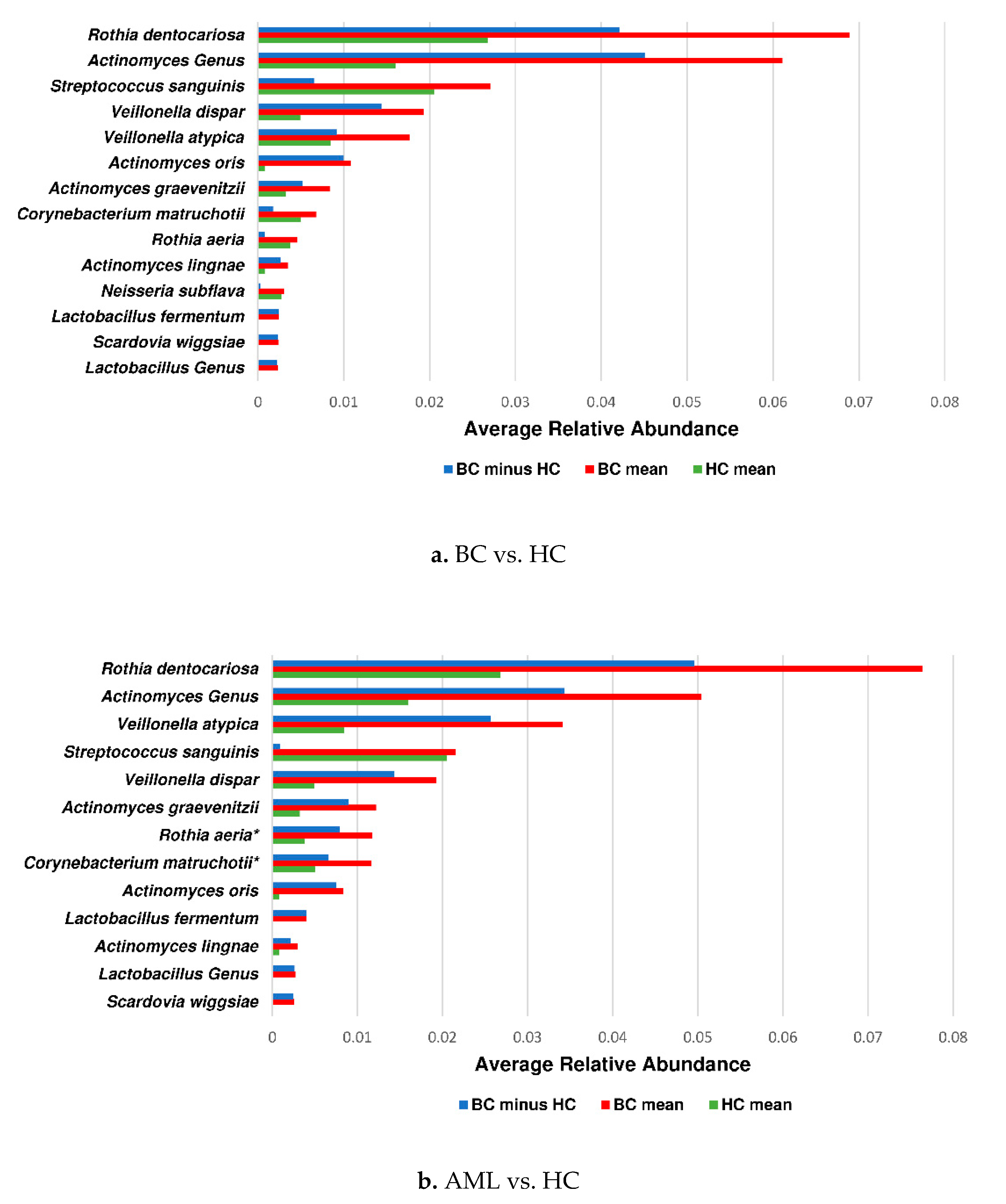
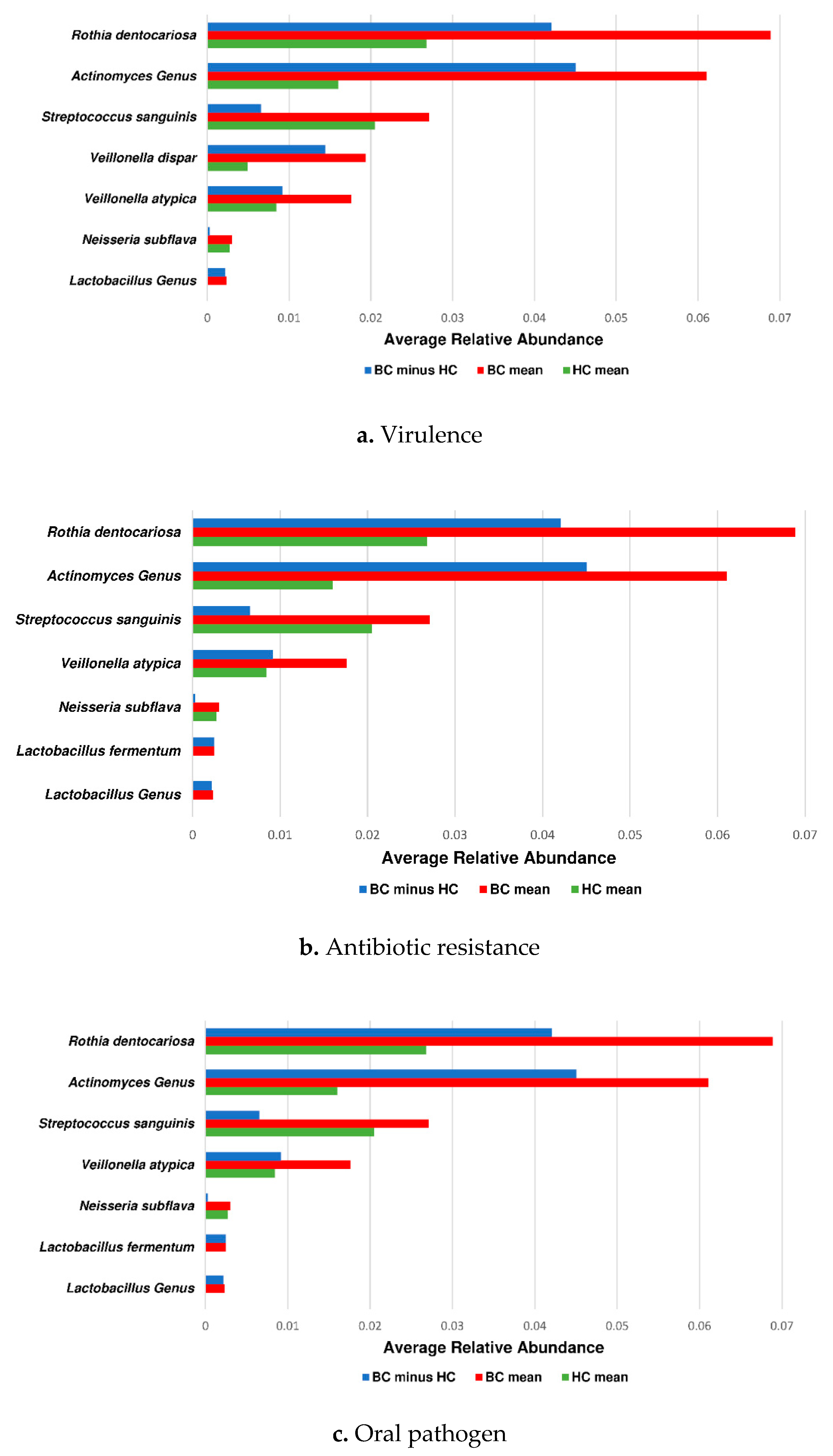
3.3. Actinomyces Genus and Rothia Dentocariosa Account for a Large Difference between the BC and HC Groups
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Montagnana, M.; Lippi, G. Cancer diagnostics: Current concepts and future perspectives. Ann. Transl. Med. 2017, 5, 268. [Google Scholar] [CrossRef] [Green Version]
- Pucci, C.; Martinelli, C.; Ciofani, G. Innovative approaches for cancer treatment: Current perspectives and new challenges. Ecancermedicalscience 2019, 13, 961. [Google Scholar] [CrossRef]
- Karpinski, T.M. Role of Oral Microbiota in Cancer Development. Microorganisms 2019, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, H.; Varoni, E.M.; Cochis, A.; Cordaro, M.; Gallenzi, P.; Patini, R.; Staderini, E.; Lajolo, C.; Rimondini, L.; Rocchetti, V. Oral Dysbiosis in Pancreatic Cancer and Liver Cirrhosis: A Review of the Literature. Biomedicines 2018, 6, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, J.J.; Zhang, L.; Zhou, H.; Chia, D.; Elashoff, D.; Akin, D.; Paster, B.J.; Joshipura, K.; Wong, D.T. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut 2012, 61, 582–588. [Google Scholar] [CrossRef]
- Mitsuhashi, K.; Nosho, K.; Sukawa, Y.; Matsunaga, Y.; Ito, M.; Kurihara, H.; Kanno, S.; Igarashi, H.; Naito, T.; Adachi, Y.; et al. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget 2015, 6, 7209–7220. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum—Symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef]
- Klimesova, K.; Jiraskova Zakostelska, Z.; Tlaskalova-Hogenova, H. Oral Bacterial and Fungal Microbiome Impacts Colorectal Carcinogenesis. Front. Microbiol. 2018, 9, 774. [Google Scholar] [CrossRef]
- Hu, J.; Han, S.; Chen, Y.; Ji, Z. Variations of Tongue Coating Microbiota in Patients with Gastric Cancer. Biomed. Res. Int. 2015, 2015, 173729. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Winckler, B.; Lu, M.; Cheng, H.; Yuan, Z.; Yang, Y.; Jin, L.; Ye, W. Oral Microbiota and Risk for Esophageal Squamous Cell Carcinoma in a High-Risk Area of China. PLoS ONE 2015, 10, e0143603. [Google Scholar] [CrossRef] [PubMed]
- Chua, L.L.; Rajasuriar, R.; Azanan, M.S.; Abdullah, N.K.; Tang, M.S.; Lee, S.C.; Woo, Y.L.; Lim, Y.A.; Ariffin, H.; Loke, P. Reduced microbial diversity in adult survivors of childhood acute lymphoblastic leukemia and microbial associations with increased immune activation. Microbiome 2017, 5, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xue, J.; Zhou, X.; You, M.; Du, Q.; Yang, X.; He, J.; Zou, J.; Cheng, L.; Li, M.; et al. Oral microbiota distinguishes acute lymphoblastic leukemia pediatric hosts from healthy populations. PLoS ONE 2014, 9, e102116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Miao, R.; Zhu, Y.; Lin, C.; Yang, X.; Jia, R.; Linghan, K.; Wan, C.; Deng, J. A new insight into acute lymphoblastic leukemia in children: Influences of changed intestinal microfloras. BMC Pediatr. 2020, 20, 290. [Google Scholar] [CrossRef]
- Galloway-Pena, J.R.; Smith, D.P.; Sahasrabhojane, P.; Wadsworth, W.D.; Fellman, B.M.; Ajami, N.J.; Shpall, E.J.; Daver, N.; Guindani, M.; Petrosino, J.F.; et al. Characterization of oral and gut microbiome temporal variability in hospitalized cancer patients. Genome Med. 2017, 9, 21. [Google Scholar] [CrossRef] [Green Version]
- Ames, N.J.; Sulima, P.; Ngo, T.; Barb, J.; Munson, P.J.; Paster, B.J.; Hart, T.C. A characterization of the oral microbiome in allogeneic stem cell transplant patients. PLoS ONE 2012, 7, e47628. [Google Scholar] [CrossRef]
- Mougeot, J.C.; Beckman, M.F.; Stevens, C.B.; Almon, K.G.; Morton, D.S.; Von Bultzingslowen, I.; Brennan, M.T.; Mougeot, F.B. Lasting Gammaproteobacteria profile changes characterized hematological cancer patients who developed oral mucositis following conditioning therapy. J. Oral Microbiol. 2020, 12, 1761135. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosikowska, U.; Rybojad, P.; Stepien-Pysniak, D.; Zbikowska, A.; Malm, A. Changes in the prevalence and biofilm formation of Haemophilus influenzae and Haemophilus parainfluenzae from the respiratory microbiota of patients with sarcoidosis. BMC Infect. Dis. 2016, 16, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Lu, W.; Tu, Q.; Ge, Y.; He, J.; Zhou, Y.; Gou, Y.; Van Nostrand, J.D.; Qin, Y.; Li, J.; et al. Preliminary analysis of salivary microbiome and their potential roles in oral lichen planus. Sci. Rep. 2016, 6, 22943. [Google Scholar] [CrossRef] [PubMed]
- Valour, F.; Senechal, A.; Dupieux, C.; Karsenty, J.; Lustig, S.; Breton, P.; Gleizal, A.; Boussel, L.; Laurent, F.; Braun, E.; et al. Actinomycosis: Etiology, clinical features, diagnosis, treatment, and management. Infect. Drug Resist. 2014, 7, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Roh, Y.H.; Park, K.J.; Byun, K.D.; Roh, M.S.; Choi, H.J. Abdominal actinomycosis misconceived as intestinal lymphoma: Report of a case. Int. J. Surg. Case Rep. 2019, 60, 171–174. [Google Scholar] [CrossRef]
- Savoca, E.; Mehra, S.; Waldman, E.H. A case of pediatric cervicofacial actinomyces masquerading as malignancy: Case report and review of the literature. Int. J. Pediatr. Otorhinolaryngol. 2019, 116, 204–208. [Google Scholar] [CrossRef]
- von Graevenitz, A. Rothia dentocariosa: Taxonomy and differential diagnosis. Clin. Microbiol. Infect. 2004, 10, 399–402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Kong, C.; Yang, Y.; Cai, S.; Li, X.; Cai, G.; Ma, Y. Human oral microbiome dysbiosis as a novel non-invasive biomarker in detection of colorectal cancer. Theranostics 2020, 10, 11595–11606. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. CA Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.J.; Rawls, J.F.; Randall, T.; Burcal, L.; Mpande, C.N.; Jenkins, N.; Jovov, B.; Abdo, Z.; Sandler, R.S.; Keku, T.O. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes 2010, 1, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Clarridge, J.E., 3rd; Zhang, Q. Genotypic diversity of clinical Actinomyces species: Phenotype, source, and disease correlation among genospecies. J. Clin. Microbiol. 2002, 40, 3442–3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, X.; Zhu, Y.; Ouyang, J.; Wang, Y.; Lei, Q.; Xia, J.; Guan, Y.; Zhang, J.; Guo, J.; He, Y.; et al. Alterations of gut microbiome accelerate multiple myeloma progression by increasing the relative abundances of nitrogen-recycling bacteria. Microbiome 2020, 8, 74. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | BC | HC | AML | |||||
|---|---|---|---|---|---|---|---|---|
| (N = 39) | (N = 27) | (N = 16) | ||||||
| Age yrs (mean (SD)) * | 52.2 (15.08) | 53.2 (14.07) | 53.2 (14.97) | |||||
| Age range yrs | 25–76 | 24–84 | 27–76 | |||||
| Gender (M/F) & | 15/24 | 7/19 | 9/7 | |||||
| Ethnicity $ | ||||||||
| Caucasian | 24 | 24 | 10 | |||||
| Black | 15 | 2 | 6 | |||||
| AB treatment | 22 | 0 | 10 | |||||
| Sample site combinations | ||||||||
| BPST (n = 132) | 13 | 20 | ||||||
| BST (n = 24) | 4 | 3 | 2 | |||||
| PST (n = 57) | 16 | 3 | 7 | |||||
| ST (n = 12) | 6 | 1 | 3 | |||||
| Total # of samples | n = 124 | n = 100 | n = 49 | |||||
| PERMANOVA p-Value a,* | |||
|---|---|---|---|
| AN1 b | Group a,d | Site a,e | AB-DIA a,f |
| BPST c | 0.0008 | 0.0023 | 0.0001 |
| HC vs. BC-No AB d | 0.0004 | 0.0003 | 0.1113 |
| HC vs. BC-AB e | 0.0036 | 0.6155 | 0.0001 |
| PST f | 0.0002 | 0.00001 | 0.00001 |
| HC vs. BC-No AB g | 0.0001 | 0.0001 | 0.1544 |
| HC vs. BC-AB h | 0.0032 | 0.0182 | 0.0002 |
| HC vs. BC-AML, LYM, MM i | 0.0001 | 0.0001 | 0.1401 |
| BST j | 0.0015 | 0.0056 | 0.0171 |
| HC vs. BC-No AB k | 0.005 | 0.0031 | 0.2208 |
| HC vs. BC-AB l | 0.0044 | 0.5188 | 0.0004 |
| ST m | 0.0043 | 0.9524 | 0.0747 |
| HC vs. BC-No AB n | 0.0107 | 0.0579 | 0.1245 |
| HC vs. BC-AB o | 0.0017 | 0.3894 | 0.0083 |
| BPST cumulative p | 0.1978 | ND | 0.0023 |
| HC vs. BC-No AB q | 0.0909 | ND | 0.2512 |
| HC vs. BC-AB r | 0.4825 | ND | 0.0012 |
| PST cumulative s | 0.0857 | ND | 0.0028 |
| HC vs. BC-No AB t | 0.04 | ND | 0.2687 |
| HC vs. BC-AB u | 0.2548 | ND | 0.0026 |
| HC vs. BC- AML, LYM, MM v | 0.0039 | ND | 0.2074 |
| ST cumulative w | 0.0666 | ND | 0.1244 |
| HC vs. BC-No AB x | 0.528 | ND | 0.0136 |
| HC vs. BC-AB y | 0.0557 | ND | 0.2245 |
| AN2 z | Group c | Site d | AB-DIA e |
| HC vs. AML a,a | 0.0002 | 0.0002 | 0.1087 |
| HC vs. NAML a,b | 0.0001 | ND | ND |
| HC vs. YAML a,c | 0.0001 | ND | ND |
| Species/Genus a | Virulence b | Source c | PMID d | ABR e | Source f | Accession IDs g | Oral Pathogen h | PMID i |
|---|---|---|---|---|---|---|---|---|
| Actinomyces | Yes | PATRIC_VF; Victors | 10456927; 14600232; 1500984 | Yes | NDARO; CARD | WP_000804064.1; WP_002586627.1; WP_063856422.1; WP_000691759.1; WP_000027050.1 | Yes | 30225251 23673380 |
| Actinomyces graevenitzii | No | No | ND | |||||
| Actinomyces lingnae | ND | ND | ND | |||||
| Actinomyces oris | No | No | ND | |||||
| Corynebacterium matruchotii | No | No | ND | |||||
| Lactobacillus | Yes | VFDB; Victors; PATRIC_VF | 12207705; 15937179; 14569030; 8063392; 19818015 | Yes | ARDB; NDARO; CARD; | WP_002352254.1; WP_001038795.1; WP_002328813.1; ABI81768.1; WP_000027050.1 | Yes | 25758458 |
| Lactobacillus fermentum | No | Yes | NDARO; CARD | AAF86220.1; WP_002328813.1; WP_011100845.1 | Yes | 19088910 | ||
| Neisseria subflava | Yes | Victors; VFDB; | 11062540; 19481311; 16988225; 19050914; 18680551 | Yes | NDARO; CARD | WP_063856397.1; CAD09800.1; WP_000027057.1; AAL59753.1; WP_000480968.1 | Yes | 12324342 |
| Rothia aeria | No | No | Yes | 24951810 | ||||
| Rothia dentocariosa | Yes | Victors | 20485570; 9383163; | Yes | NDARO; CARD | CAJ67339.1; WP_000691727.1 | Yes | 27303245 |
| Scardovia inopinata | No | No | Yes | 32401932 | ||||
| Scardovia wiggsiae | No | No | Yes | 29104444 | ||||
| Streptococcus sanguinis | Yes | Victors; PATRIC_VF | 8820650; 12207705; 15731074; 10768978; 10998175; | Yes | NDARO; CARD | CAA45935.1; WP_000420317.1; WP_000420313.1; WP_000417519.1; WP_000691736.1 | Yes | 32082276 |
| Veillonella atypica | Yes | Victors | 11062540 | Yes | NDARO; CARD | WP_000018329.1; CAA45935.1; WP_001038790.1; WP_000196697.1; | Yes | 28473967 |
| Veillonella dispar | Yes | Victors | 11062540; 12207705 | No | Yes | 28473967 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mougeot, J.-L.C.; Beckman, M.F.; Langdon, H.C.; Brennan, M.T.; Bahrani Mougeot, F. Oral Microbiome Signatures in Hematological Cancers Reveal Predominance of Actinomyces and Rothia Species. J. Clin. Med. 2020, 9, 4068. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9124068
Mougeot J-LC, Beckman MF, Langdon HC, Brennan MT, Bahrani Mougeot F. Oral Microbiome Signatures in Hematological Cancers Reveal Predominance of Actinomyces and Rothia Species. Journal of Clinical Medicine. 2020; 9(12):4068. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9124068
Chicago/Turabian StyleMougeot, Jean-Luc C., Micaela F. Beckman, Holden C. Langdon, Michael T. Brennan, and Farah Bahrani Mougeot. 2020. "Oral Microbiome Signatures in Hematological Cancers Reveal Predominance of Actinomyces and Rothia Species" Journal of Clinical Medicine 9, no. 12: 4068. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9124068