Chronic Oral Palmitoylethanolamide Administration Rescues Cognitive Deficit and Reduces Neuroinflammation, Oxidative Stress, and Glutamate Levels in A Transgenic Murine Model of Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Pharmacokinetic Studies
2.2.1. Single Oral Administration of Um-PEA
2.2.2. Sub-Chronic Oral Administration of Um-PEA
2.3. Effects of A Chronic (3 Months) Treatment with Um-PEA on Cognitive Performance and Biochemical Parameters
2.3.1. Animal Treatment
2.3.2. Behavioral Test: Novel Object Recognition Test
2.3.3. Biochemical Analyses
TNF-α and IL-16 levels
Measurement of Reactive Oxygen Species
Synaptophysin Levels
Extracellular Glutamate Levels in Mouse Hippocampus
2.4. Statistical Analysis
3. Results
3.1. Basal Levels of PEA in Plasma and Brain Homogenates of Non-Tg and 3×Tg-AD Mice
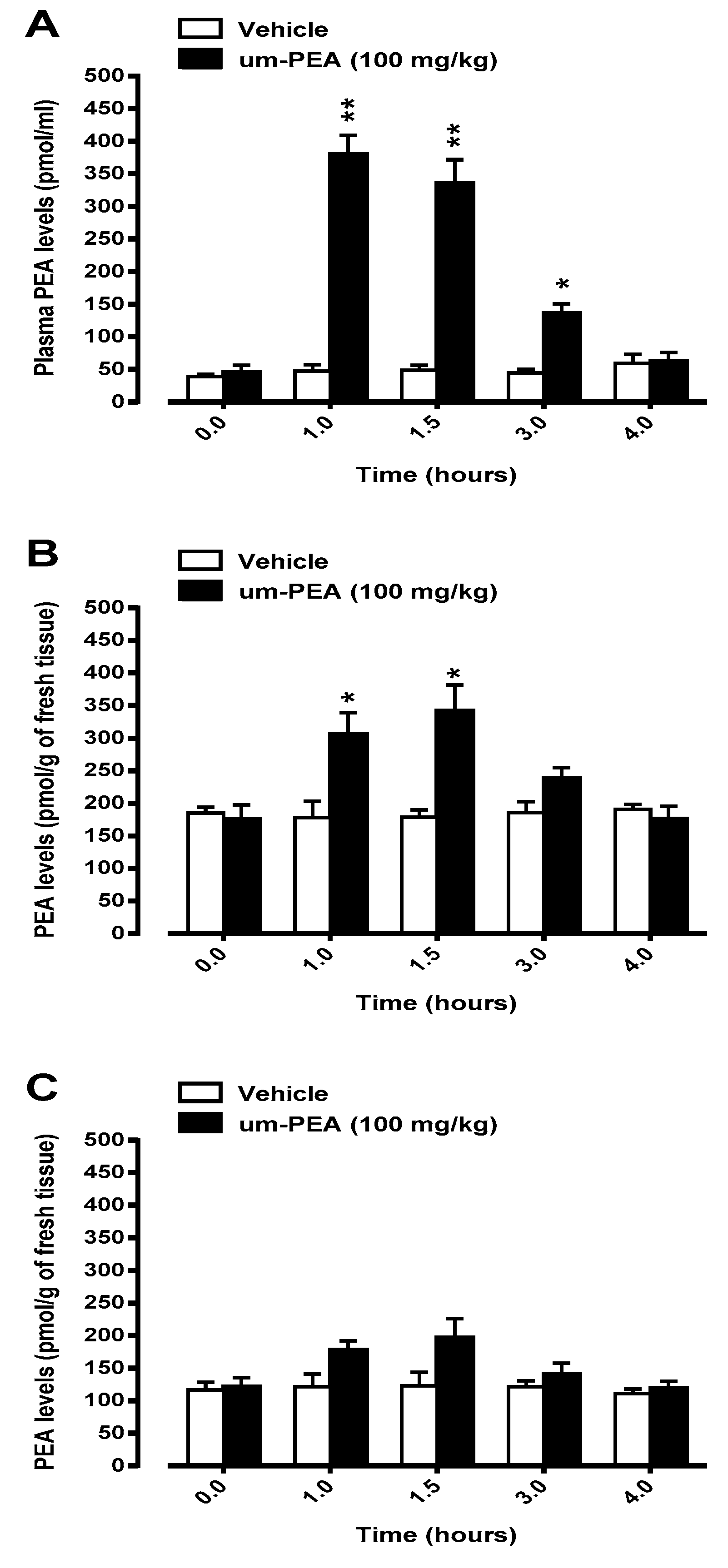
3.2. Levels of PEA in Plasma and Brain Homogenates of Non-Tg Mice after A Single Oral Administration of Um-PEA
3.3. Levels of PEA in Plasma and Brain Homogenates of Non-Tg Mice after A Sub-Chronic Oral Administration of Um-PEA
3.4. Effects of A Chronic (3 Months) Treatment with Um-PEA on Cognitive Performance and Biochemical Parameters in Non-Tg and 3×Tg-AD Mice
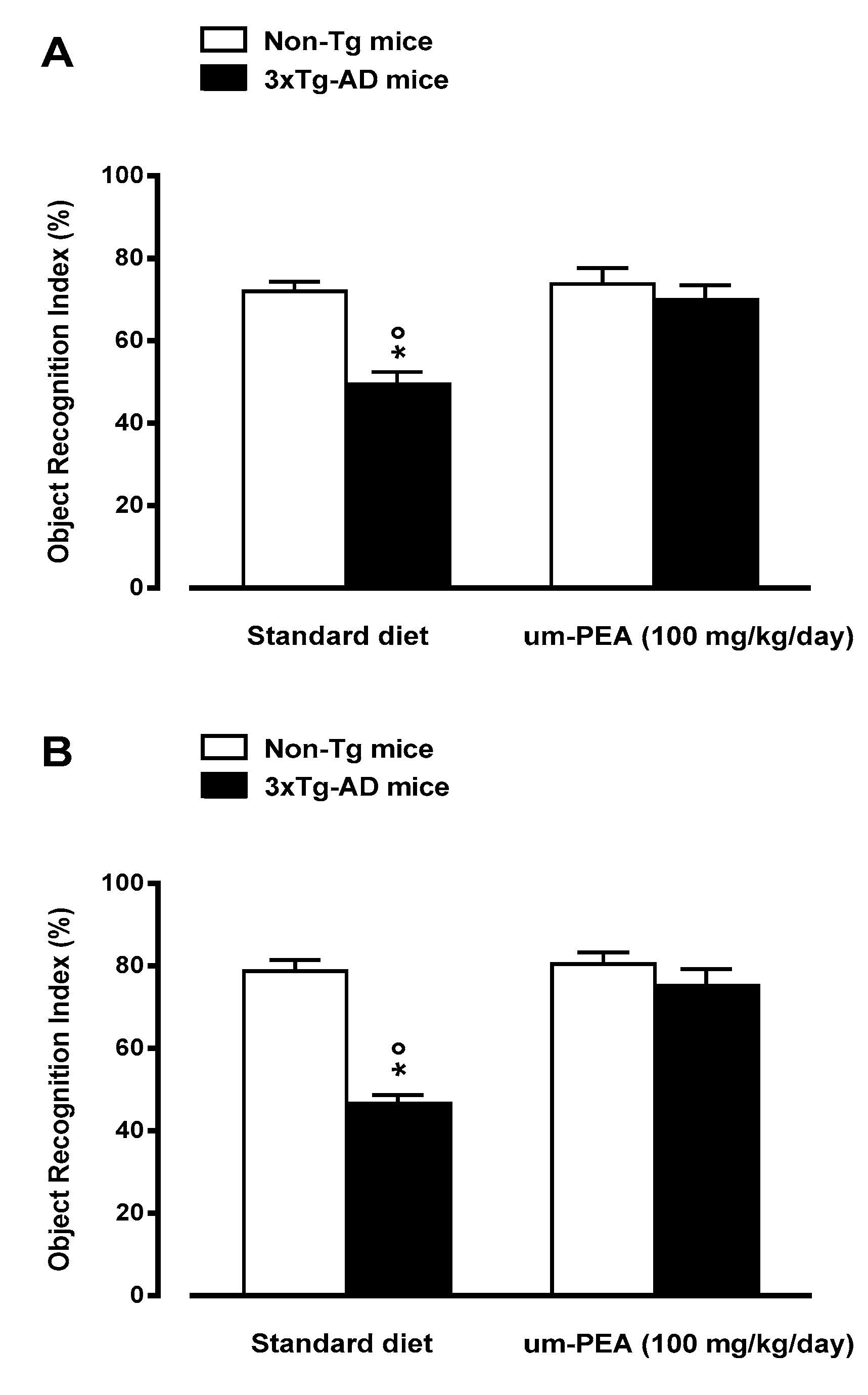
3.4.1. Um-PEA Improves learning and Memory in 5-Month-old 3×Tg-AD Mice
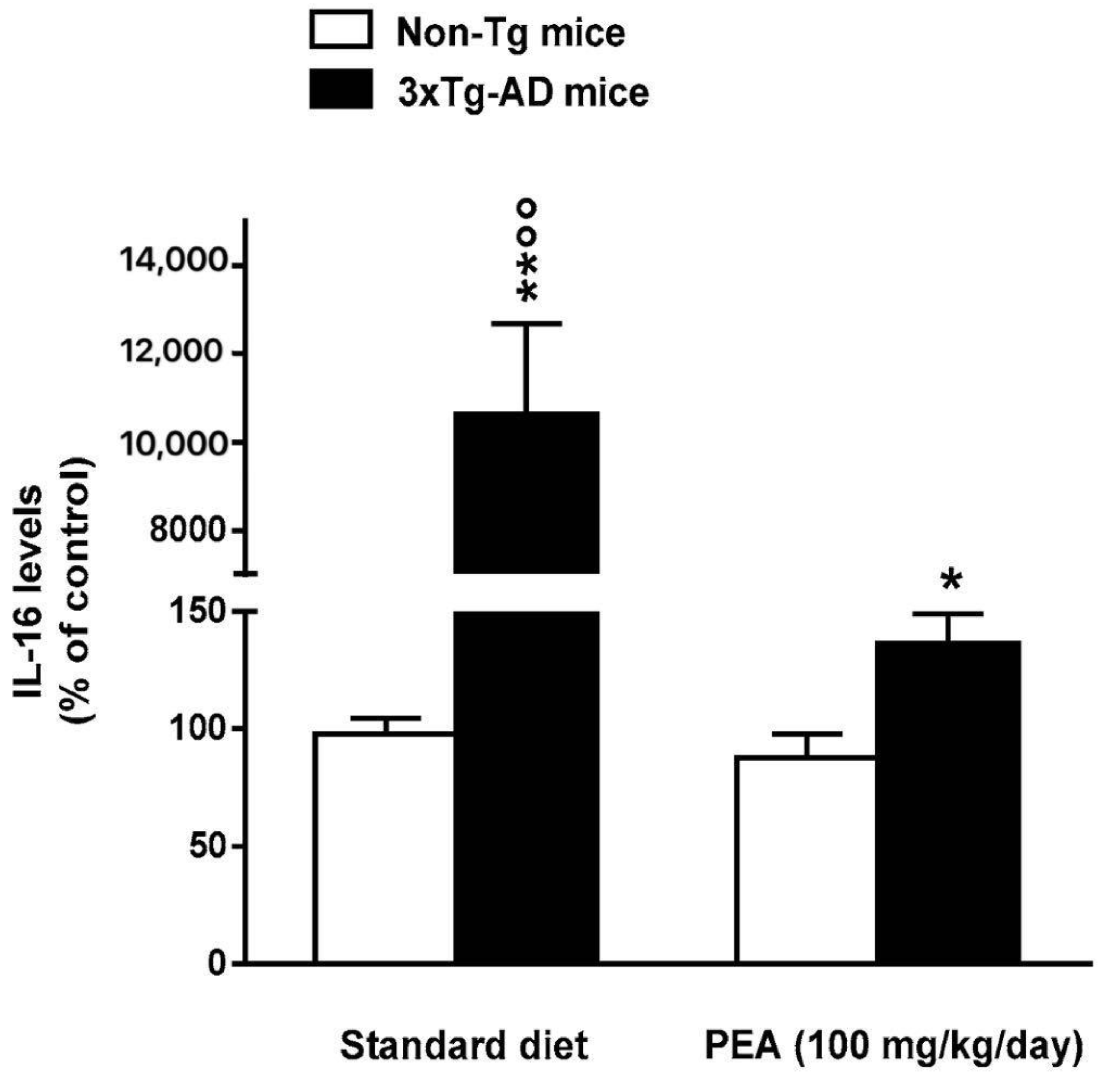
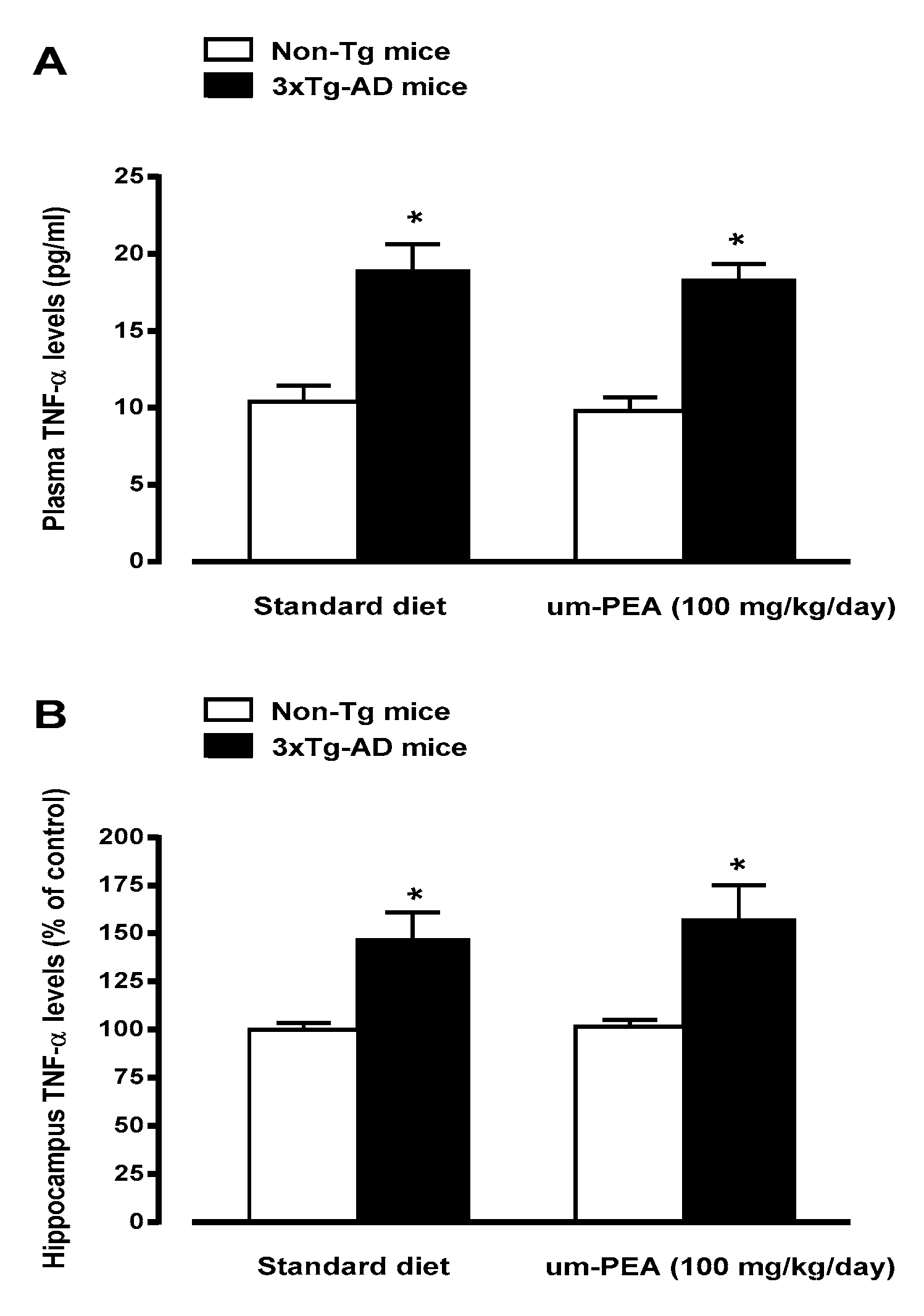
3.4.2. Um-PEA Partially Restrains Neuroinflammation
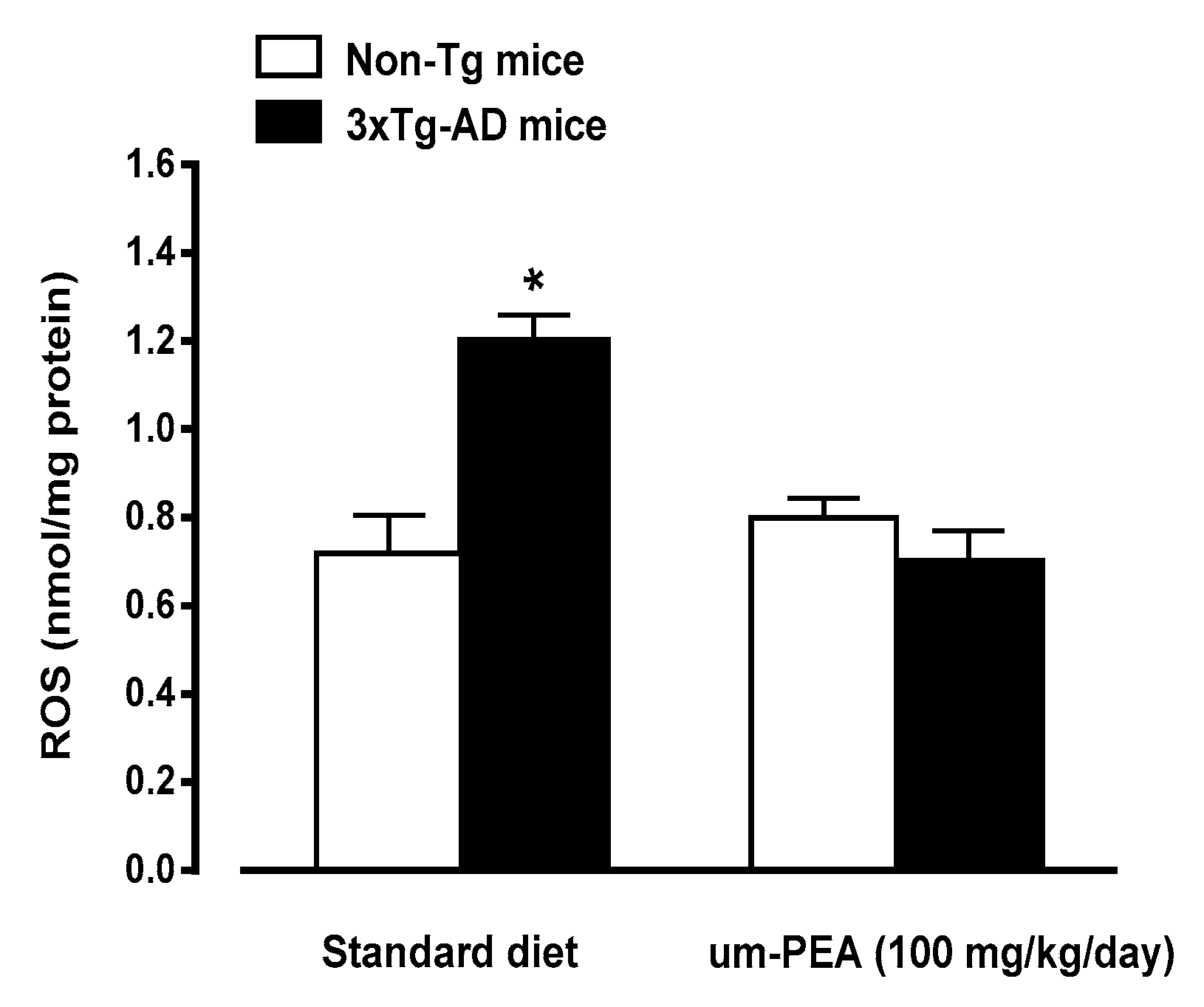
3.4.3. Um-PEA Reduces ROS Production
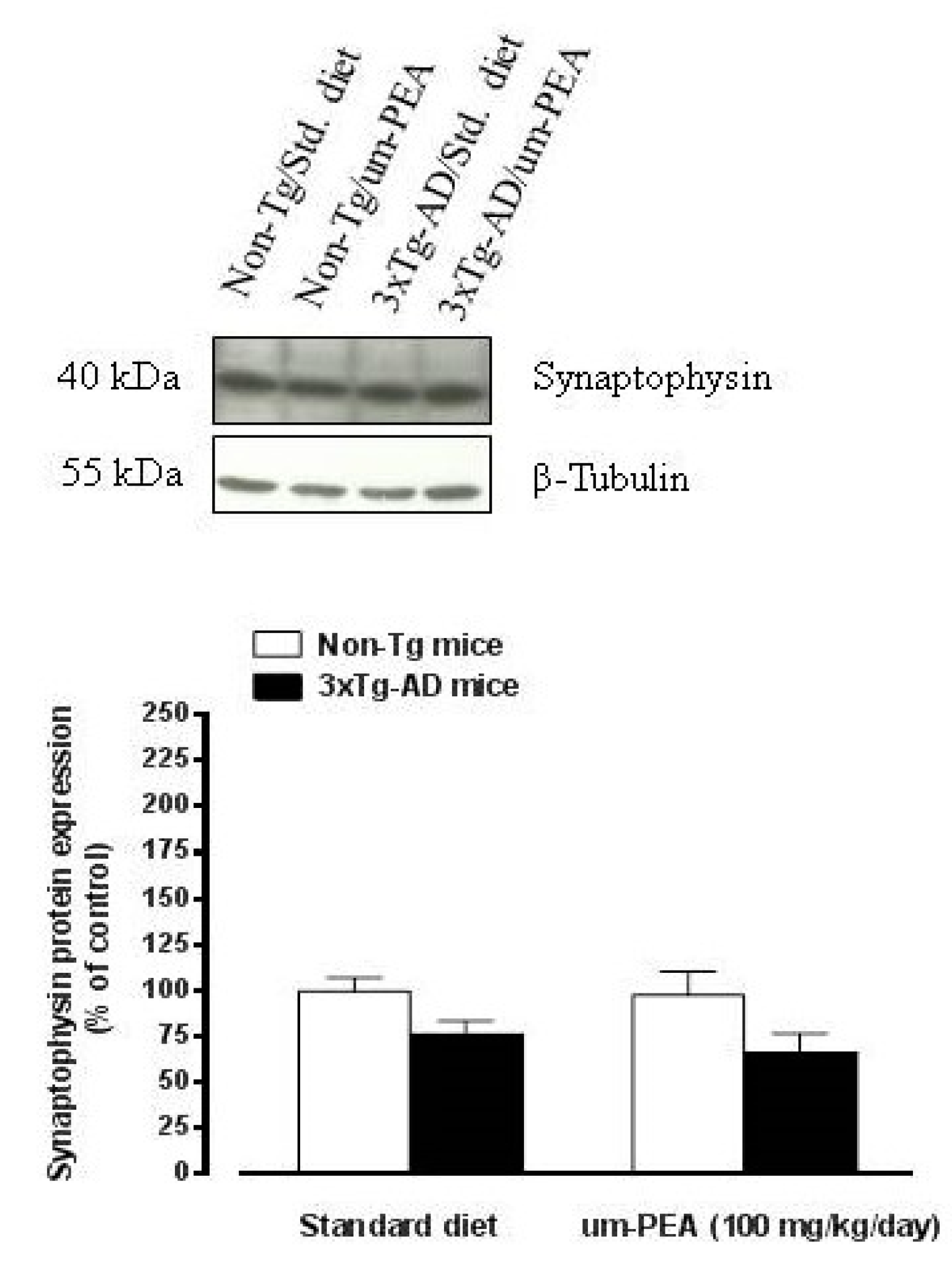
3.4.4. Um-PEA did not Affect Synaptophysin Levels in the Hippocampus of 3×Tg-AD Mice
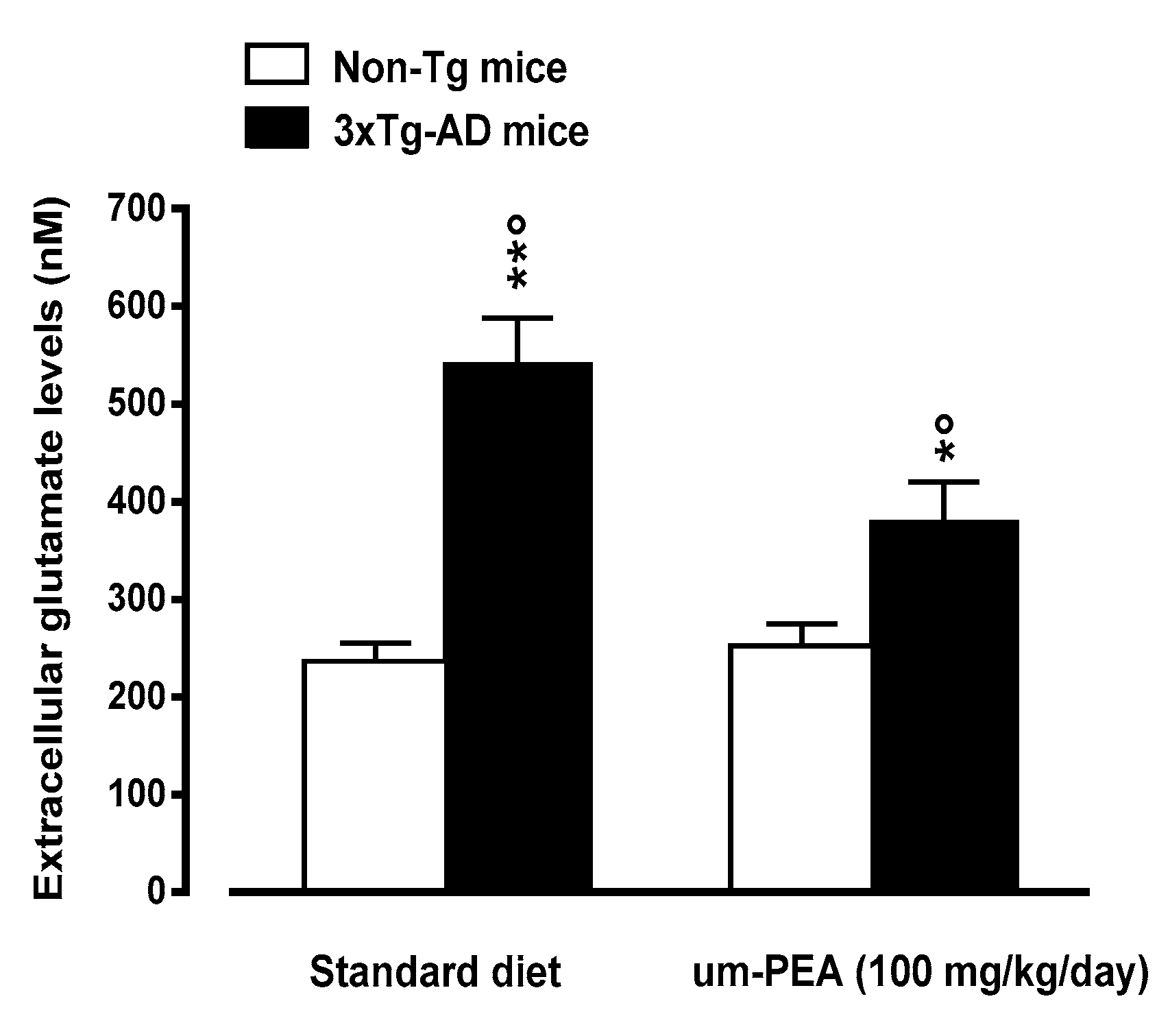
3.4.5. Um-PEA Rescues Increased Glutamate Levels in the Hippocampus of 3×Tg-AD Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Cortés, N.; Andrade, V.; Maccioni, R.B. Behavioral and Neuropsychiatric Disorders in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 63, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zvěřová, M. Clinical aspects of Alzheimer’s disease. Clin. Biochem. 2019, 72, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Edison, P.; Donat, C.K.; Sastre, M. In vivo Imaging of Glial Activation in Alzheimer’s Disease. Front. Neurol. 2018, 9, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, D.A.; Boyd-Kimball, D. Redox proteomics and amyloid β-peptide: Insights into Alzheimer disease. J. Neurochem. 2019, 151, 459–487. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Chaney, A.; Williams, S.R.; Boutin, H. In vivo molecular imaging of neuroinflammation in Alzheimer’s disease. J. Neurochem. 2019, 149, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Ameen-Ali, K.E.; Wharton, S.B.; Simpson, J.E.; Heath, P.R.; Sharp, P.; Berwick, J. Review: Neuropathology and behavioural features of transgenic murine models of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 553–570. [Google Scholar] [CrossRef] [Green Version]
- Takashi, S.; Takaomi, C.S. Neuroinflammation in mouse models of Alzheimer’s disease. Clin. Exp. Neuroimmunol. 2018, 9, 211–218. [Google Scholar]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L.; Scuderi, C. Astrocyte: An Innovative Approach for Alzheimer’s Disease Therapy. Curr. Pharm. Des. 2017, 23, 4979–4989. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Koulakoff, A.; Giaume, C. Astroglial Connexins as a Therapeutic Target for Alzheimer’s Disease. Curr. Pharm. Des. 2017, 23, 4958–4968. [Google Scholar]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Di Marzo, V. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br. J. Pharmacol. 2017, 174, 1349–1365. [Google Scholar] [CrossRef]
- Beggiato, S.; Tomasini, M.C.; Ferraro, L. Palmitoylethanolamide (PEA) as a Potential Therapeutic Agent in Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 821. [Google Scholar] [CrossRef] [Green Version]
- Tomasini, M.C.; Borelli, A.C.; Beggiato, S.; Ferraro, L.; Cassano, T.; Tanganelli, S.; Antonelli, T. Differential Effects of Palmitoylethanolamide against Amyloid-β Induced Toxicity in Cortical Neuronal and Astrocytic Primary Cultures from Wild-Type and 3xTg-AD Mice. J. Alzheimers Dis. 2015, 46, 407–421. [Google Scholar] [CrossRef]
- Beggiato, S.; Borelli, A.C.; Ferraro, L.; Tanganelli, S.; Antonelli, T.; Tomasini, M.C. Palmitoylethanolamide Blunts Amyloid-β42-Induced Astrocyte Activation and Improves Neuronal Survival in Primary Mouse Cortical Astrocyte-Neuron Co-Cultures. J. Alzheimers Dis. 2018, 61, 389–399. [Google Scholar] [CrossRef]
- Scuderi, C.; Esposito, G.; Blasio, A.; Valenza, M.; Arietti, P.; Steardo L., Jr.; Carnuccio, R.; De Filippis, D.; Petrosino, S.; Iuvone, T.; et al. Palmitoylethanolamide counteracts reactive astrogliosis induced by β-amyloid peptide. J. Cell. Mol. Med. 2011, 15, 2664–2674. [Google Scholar] [CrossRef] [Green Version]
- Bronzuoli, M.R.; Facchinetti, R.; Valenza, M.; Cassano, T.; Steardo, L.; Scuderi, C. Astrocyte Function Is Affected by Aging and Not Alzheimer’s Disease: A Preliminary Investigation in Hippocampi of 3xTg-AD Mice. Front. Pharmacol. 2019, 10, 644. [Google Scholar] [CrossRef] [Green Version]
- Impellizzeri, D.; Bruschetta, G.; Cordaro, M.; Crupi, R.; Siracusa, R.; Esposito, E.; Cuzzocrea, S. Micronized/ultramicronized palmitoylethanolamide displays superior oral efficacy compared to nonmicronized palmitoylethanolamide in a rat model of inflammatory pain. J. Neuroinflammation 2014, 11, 136. [Google Scholar] [CrossRef] [Green Version]
- Petrosino, S.; Cordaro, M.; Verde, R.; Schiano Moriello, A.; Marcolongo, G.; Schievano, C.; Siracusa, R.; Piscitelli, F.; Peritore, A.F.; Crupi, R.; et al. Oral Ultramicronized Palmitoylethanolamide: Plasma and Tissue Levels and Spinal Anti-hyperalgesic Effect. Front. Pharmacol. 2018, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Bronzuoli, M.R.; Facchinetti, R.; Pace, L.; Ferraro, L.; Broad, K.D.; Serviddio, G.; Bellanti, F.; Palombelli, G.; Carpinelli, G.; et al. Ultramicronized palmitoylethanolamide rescues learning and memory impairments in a triple transgenic mouse model of Alzheimer’s disease by exerting anti-inflammatory and neuroprotective effects. Transl. Psychiatry 2018, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Bilkei-Gorzo, A. Genetic mouse models of brain ageing and Alzheimer’s disease. Pharmacol. Ther. 2014, 142, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Onos, K.D.; Sukoff Rizzo, S.J.; Howell, G.R.; Sasner, M. Toward more predictive genetic mouse models of Alzheimer’s disease. Brain Res. Bull. 2016, 122, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef] [Green Version]
- Resende, R.; Moreira, P.I.; Proença, T.; Deshpande, A.; Busciglio, J.; Pereira, C.; Oliveira, C.R. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic. Biol. Med. 2008, 44, 2051–2057. [Google Scholar] [CrossRef] [Green Version]
- Young, M.L.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol. Cell. Neurosci. 2019, 101, 103409. [Google Scholar] [CrossRef]
- Cassano, T.; Serviddio, G.; Gaetani, S.; Romano, A.; Dipasquale, P.; Cianci, S.; Bellanti, F.; Laconca, L.; Romano, A.D.; Padalino, I.; et al. Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol. Aging 2012, 33, 1121.e1–1121.e12. [Google Scholar] [CrossRef]
- Sharma, K.; Singh, R.R.; Kandaswamy, M.; Mithra, C.; Giri, S.; Rajagopal, S.; Mullangi, R. LC-MS/MS-ESI method for simultaneous quantitation of three endocannabinoids and its application to rat pharmacokinetic studies. Bioanalysis 2011, 3, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Liput, D.J.; Tsakalozou, E.; Hammell, D.C.; Paudel, K.S.; Nixon, K.; Stinchcomb, A.L. Quantification of anandamide, oleoylethanolamide and palmitoylethanolamide in rodent brain tissue using high performance liquid chromatography-electrospray mass spectroscopy. J. Pharm. Anal. 2014, 4, 234–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.W.; Kim, S.J.; Kim, M.S. Oxidative stress with tau hyperphosphorylation in memory impaired 1,2-diacetylbenzene-treated mice. Toxicol. Lett. 2017, 279, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Beggiato, S.; Giuliani, A.; Sivilia, S.; Lorenzini, L.; Antonelli, T.; Imbimbo, B.P.; Giardino, L.; Calzà, L.; Ferraro, L. CHF5074 and LY450139 sub-acute treatments differently affect cortical extracellular glutamate levels in pre-plaque Tg2576 mice. Neuroscience 2014, 266, 13–22. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Vacondio, F.; Bassi, M.; Silva, C.; Castelli, R.; Carmi, C.; Scalvini, L.; Lodola, A.; Vivo, V.; Flammini, L.; Barocelli, E.; et al. Amino Acid Derivatives as Palmitoylethanolamide Prodrugs: Synthesis, In Vitro Metabolism and In Vivo Plasma Profile in Rats. PLoS ONE 2015, 10, e0128699. [Google Scholar] [CrossRef] [Green Version]
- Grillo, S.L.; Keereetaweep, J.; Grillo, M.A.; Chapman, K.D.; Koulen, P. N-Palmitoylethanolamine depot injection increased its tissue levels and those of other acylethanolamide lipids. Drug Des. Devel. Ther. 2013, 7, 747–752. [Google Scholar]
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide protects against the amyloid-β25-35-induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology 2012, 37, 1784–1792. [Google Scholar] [CrossRef] [Green Version]
- Scuderi, C.; Stecca, C.; Valenza, M.; Ratano, P.; Bronzuoli, M.R.; Bartoli, S.; Steardo, L.; Pompili, E.; Fumagalli, L.; Campolongo, P.; et al. Palmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1419. [Google Scholar] [CrossRef] [Green Version]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [Green Version]
- Anvar, N.E.; Saliminejad, K.; Ohadi, M.; Kamali, K.; Daneshmand, P.; Khorshid, H.R. Association between polymorphisms in Interleukin-16 gene and risk of late-onset Alzheimer’s disease. J. Neurol. Sci. 2015, 358, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Gugliandolo, E.; Fusco, R.; Biundo, F.; D’Amico, R.; Benedetto, F.; Di Paola, R.; Cuzzocrea, S. Palmitoylethanolamide and Polydatin combination reduces inflammation and oxidative stress in vascular injury. Pharmacol. Res. 2017, 123, 83–92. [Google Scholar] [CrossRef]
- Li, M.; Wang, D.; Bi, W.; Jiang, Z.E.; Piao, R.; Yu, H. N-Palmitoylethanolamide Exerts Antidepressant-Like Effects in Rats: Involvement of PPARα Pathway in the Hippocampus. J. Pharmacol. Exp. Ther. 2019, 369, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Impellizzeri, D.; Peritore, A.F.; Cordaro, M.; Gugliandolo, E.; Siracusa, R.; Crupi, R.; D’Amico, R.; Fusco, R.; Evangelista, M.; Cuzzocrea, S.; et al. The neuroprotective effects of micronized PEA (PEA-m) formulation on diabetic peripheral neuropathy in mice. FASEB J. 2019, 33, 11364–11380. [Google Scholar] [CrossRef]
- Zolese, G.; Bacchetti, T.; Masciangelo, S.; Ragni, L.; Ambrosi, S.; Ambrosini, A.; Marini, M.; Ferretti, G. Effect of acylethanolamides on lipid peroxidation and paraoxonase activity. Biofactors 2008, 33, 201–209. [Google Scholar] [CrossRef]
- Nakajima, A.; Aoyama, Y.; Shin, E.J.; Nam, Y.; Kim, H.C.; Nagai, T.; Yokosuka, A.; Mimaki, Y.; Yokoi, T.; Ohizumi, Y.; et al. Nobiletin, a citrus flavonoid, improves cognitive impairment and reduces soluble Aβ levels in a triple transgenic mouse model of Alzheimer’s disease (3XTg-AD). Behav. Brain Res. 2015, 289, 69–77. [Google Scholar] [CrossRef]
- Shen, L.; Chen, Y.; Yang, A.; Chen, C.; Liao, L.; Li, S.; Ying, M.; Tian, J.; Liu, Q.; Ni, J. Redox Proteomic Profiling of Specifically Carbonylated Proteins in the Serum of Triple Transgenic Alzheimer’s Disease Mice. Int. J. Mol. Sci. 2016, 17, 469. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Poddighe, L.; Serra, M.P.; Boi, M.; Melis, T.; Lisai, S.; Murru, E.; Muredda, L.; Collu, M.; Banni, S.; et al. Preventive Effects of Resveratrol on Endocannabinoid System and Synaptic Protein Modifications in Rat Cerebral Cortex Challenged by Bilateral Common Carotid Artery Occlusion and Reperfusion. Int. J. Mol. Sci. 2018, 19, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raso, G.M.; Esposito, E.; Vitiello, S.; Iacono, A.; Santoro, A.; D’Agostino, G.; Sasso, O.; Russo, R.; Piazza, P.V.; Calignano, A.; et al. Palmitoylethanolamide stimulation induces allopregnanolone synthesis in C6 Cells and primary astrocytes: Involvement of peroxisome-proliferator activated receptor-α. J. Neuroendocrinol. 2011, 23, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, M.M.; Clos, M.V.; Ratia, M.; Gonzalez, D.; Lithner, C.U.; Camps, P.; Muñoz-Torrero, D.; Badia, A.; Giménez-Llort, L.; Nordberg, A. Effect of huprine X on β-amyloid, synaptophysin and α7 neuronal nicotinic acetylcholine receptors in the brain of 3xTg-AD and APPswe transgenic mice. Neurodegener. Dis. 2010, 7, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Baazaoui, N.; Flory, M.; Iqbal, K. Synaptic Compensation as a Probable Cause of Prolonged Mild Cognitive Impairment in Alzheimer’s Disease: Implications from a Transgenic Mouse Model of the Disease. J. Alzheimers Dis. 2017, 56, 1385–1401. [Google Scholar] [CrossRef] [PubMed]
- Graff-Radford, J.; Kantarci, K. Magnetic resonance spectroscopy in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2013, 9, 687–696. [Google Scholar] [PubMed] [Green Version]
- Verkhratsky, A.; Steardo, L.; Peng, L.; Parpura, V. Astroglia, glutamatergic transmission and psychiatric diseases. Adv. Neurobiol. 2016, 13, 307–326. [Google Scholar] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Boccella, S.; Cristiano, C.; Romano, R.; Iannotta, M.; Belardo, C.; Farina, A.; Guida, F.; Piscitelli, F.; Palazzo, E.; Mazzitelli, M.; et al. Ultra-micronized palmitoylethanolamide rescues the cognitive decline-associated loss of neural plasticity in the neuropathic mouse entorhinal cortex-dentate gyrus pathway. Neurobiol. Dis. 2019, 121, 106–119. [Google Scholar] [CrossRef]
- Esposito, E.; Cuzzocrea, S. Palmitoylethanolamide is a new possible pharmacological treatment for the inflammation associated with trauma. Mini Rev. Med. Chem. 2013, 13, 237–255. [Google Scholar]
- Calabrò, R.S.; Naro, A.; De Luca, R.; Leonardi, S.; Russo, M.; Marra, A.; Bramanti, P. PEALut efficacy in mild cognitive impairment: Evidence from a SPECT case study! Aging Clin. Exp. Res. 2016, 28, 1279–1282. [Google Scholar] [CrossRef]
- Brotini, S.; Schievano, C.; Guidi, L. Ultra-micronized palmitoylethanolamide: An efficacious adjuvant therapy for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2017, 16, 705–713. [Google Scholar] [CrossRef]
- Angelini, R.; Argueta, D.A.; Piomelli, D.; DiPatrizio, N.V. Identification of a Widespread Palmitoylethanolamide Contamination in Standard Laboratory Glassware. Cannabis Cannabinoid Res. 2017, 2, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Paouri, E.; Georgopoulos, S. Systemic and CNS Inflammation Crosstalk: Implications for Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsson, L.; Mattsson, S.; Fowler, C.J. Palmitoylethanolamide for the treatment of pain: Pharmacokinetics, safety and efficacy. Br. J. Clin. Pharmacol. 2016, 82, 932–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestmann, E.R. Safety of micronized palmitoylethanolamide (microPEA): Lack of toxicity and genotoxic potential. Food Sci. Nutr. 2016, 5, 292–309. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beggiato, S.; Tomasini, M.C.; Cassano, T.; Ferraro, L. Chronic Oral Palmitoylethanolamide Administration Rescues Cognitive Deficit and Reduces Neuroinflammation, Oxidative Stress, and Glutamate Levels in A Transgenic Murine Model of Alzheimer’s Disease. J. Clin. Med. 2020, 9, 428. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9020428
Beggiato S, Tomasini MC, Cassano T, Ferraro L. Chronic Oral Palmitoylethanolamide Administration Rescues Cognitive Deficit and Reduces Neuroinflammation, Oxidative Stress, and Glutamate Levels in A Transgenic Murine Model of Alzheimer’s Disease. Journal of Clinical Medicine. 2020; 9(2):428. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9020428
Chicago/Turabian StyleBeggiato, Sarah, Maria Cristina Tomasini, Tommaso Cassano, and Luca Ferraro. 2020. "Chronic Oral Palmitoylethanolamide Administration Rescues Cognitive Deficit and Reduces Neuroinflammation, Oxidative Stress, and Glutamate Levels in A Transgenic Murine Model of Alzheimer’s Disease" Journal of Clinical Medicine 9, no. 2: 428. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9020428