Inositol 1,4,5-Trisphosphate Receptors in Human Disease: A Comprehensive Update



Abstract
:1. Introduction
2. ITPRs and Neurological Disorders
2.1. Spinocerebellar Ataxia
2.2. Huntington’s Disease and Alzheimer’s Disease
2.3. Gillespie Syndrome
2.4. Autism Spectrum Disorder
2.5. Amyotrophic Lateral Sclerosis
3. ITPRs in Autoimmune Disorders
4. ITPRs and Anhidrosis
5. ITPRs and Cancer
6. Potential Role of ITPRs in Human Disease: Evidence from GWAS
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mikoshiba, K.; Changeux, J.P. Morphological and biochemical studies on isolated molecular and granular layers from bovine cerebellum. Brain Res. 1978, 142, 487–504. [Google Scholar] [CrossRef]
- Mikoshiba, K.; Huchet, M.; Changeux, J.P. Biochemical and immunological studies on the P400 protein, a protein characteristic of the Purkinje cell from mouse and rat cerebellum. Dev. Neurosci. 1979, 2, 254–275. [Google Scholar] [CrossRef] [PubMed]
- Crepel, F.; Dupont, J.L.; Gardette, R. Selective absence of calcium spikes in Purkinje cells of staggerer mutant mice in cerebellar slices maintained in vitro. J. Physiol. 1984, 346, 111–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, N.; Niinobe, M.; Nakahira, K.; Mikoshiba, K. Purification and characterization of P400 protein, a glycoprotein characteristic of Purkinje cell, from mouse cerebellum. J. Neurochem. 1988, 51, 1724–1730. [Google Scholar] [CrossRef]
- Furuichi, T.; Yoshikawa, S.; Miyawaki, A.; Wada, K.; Maeda, N.; Mikoshiba, K. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature 1989, 342, 32–38. [Google Scholar] [CrossRef]
- Furuichi, T.; Yoshikawa, S.; Mikoshiba, K. Nucleotide sequence of cDNA encoding P400 protein in the mouse cerebellum. Nucleic Acids Res. 1989, 17, 5385–5386. [Google Scholar] [CrossRef] [Green Version]
- Iino, M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. J. Gen. Physiol 1990, 95, 1103–1122. [Google Scholar] [CrossRef] [Green Version]
- Finch, E.A.; Turner, T.J.; Goldin, S.M. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science 1991, 252, 443–446. [Google Scholar] [CrossRef]
- Santulli, G.; Nakashima, R.; Yuan, Q.; Marks, A.R. Intracellular calcium release channels: An update. J. Physiol. 2017, 595, 3041–3051. [Google Scholar] [CrossRef] [Green Version]
- Paknejad, N.; Hite, R.K. Structural basis for the regulation of inositol trisphosphate receptors by Ca(2+) and IP3. Nat. Struct. Mol. Biol. 2018, 25, 660–668. [Google Scholar] [CrossRef]
- Belkacemi, A.; Hui, X.; Wardas, B.; Laschke, M.W.; Wissenbach, U.; Menger, M.D.; Lipp, P.; Beck, A.; Flockerzi, V. IP3 Receptor-Dependent Cytoplasmic Ca(2+) Signals Are Tightly Controlled by Cavbeta3. Cell Rep. 2018, 22, 1339–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rong, Y.P.; Bultynck, G.; Aromolaran, A.S.; Zhong, F.; Parys, J.B.; De Smedt, H.; Mignery, G.A.; Roderick, HL.; Bootman, M.D.; Distelhorst, C.W. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 14397–14402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prole, D.L.; Taylor, C.W. Structure and Function of IP3 Receptors. Cold Spring Harb Perspect Biol 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.C.; Baek, K.; Lu, Z. Apo and InsP(3)-bound crystal structures of the ligand-binding domain of an InsP(3) receptor. Nat. Struct Mol. Biol. 2011, 18, 1172–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, M.D.; Velamakanni, S.; Ishiyama, N.; Stathopulos, P.B.; Rossi, A.M.; Khan, S.A.; Dale, P.; Li, C.; Ames, J.B.; Ikura, M.; et al. Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 2012, 483, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Fan, G.; Baker, M.L.; Wang, Z.; Baker, M.R.; Sinyagovskiy, P.A.; Chiu, W.; Ludtke, S.J.; Serysheva, I.I. Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 2015, 527, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Chandran, A.; Chee, X.; Prole, D.L.; Rahman, T. Exploration of inositol 1,4,5-trisphosphate (IP3) regulated dynamics of N-terminal domain of IP3 receptor reveals early phase molecular events during receptor activation. Sci. Rep. 2019, 9, 2454. [Google Scholar] [CrossRef] [Green Version]
- Hamada, K.; Miyatake, H.; Terauchi, A.; Mikoshiba, K. IP3-mediated gating mechanism of the IP3 receptor revealed by mutagenesis and X-ray crystallography. Proc. Natl. Acad. Sci. USA 2017, 114, 4661–4666. [Google Scholar] [CrossRef] [Green Version]
- Lock, J.T.; Alzayady, K.J.; Yule, D.I.; Parker, I. All three IP3 receptor isoforms generate Ca(2+) puffs that display similar characteristics. Sci. Signal. 2018, 11, eaau0344. [Google Scholar] [CrossRef] [Green Version]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Chan, C.; Ooashi, N.; Akiyama, H.; Fukuda, T.; Inoue, M.; Matsu-Ura, T.; Shimogori, T.; Mikoshiba, K.; Kamiguchi, H. Inositol 1,4,5-Trisphosphate Receptor Type 3 Regulates Neuronal Growth Cone Sensitivity to Guidance Signals. iScience 2020, 23, 100963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Q.; Yang, J.; Santulli, G.; Reiken, S.R.; Wronska, A.; Kim, M.M.; Osborne, B.W.; Lacampagne, A.; Yin, Y.; Marks, A.R. Maintenance of normal blood pressure is dependent on IP3R1-mediated regulation of eNOS. Proc. Natl. Acad. Sci. USA 2016, 113, 8532–8537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.J.; Zhang, D.; Guerra, M.T.; Brill, A.L.; Goedeke, L.; Nasiri, A.R.; Rabin-Court, A.; Wang, Y.; Peng, L.; Dufour, S.; et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 2020, 579, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, G.; Goode, J.; Paz, J.C.; Ouyang, K.; Screaton, R.; Fischer, W.H.; Chen, J.; Tabas, I.; Montminy, M. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature 2012, 485, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca(2+)-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef]
- Cheung, K.H.; Mei, L.; Mak, D.O.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 2010, 3, ra22. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, J.; Trimarco, B.; Iaccarino, G.; Santulli, G. New Insights in Cardiac Calcium Handling and Excitation-Contraction Coupling. Adv. Exp. Med. Biol 2018, 1067, 373–385. [Google Scholar]
- Huang, W.; Cane, M.C.; Mukherjee, R.; Szatmary, P.; Zhang, X.; Elliott, V.; Ouyang, Y.; Chvanov, M.; Latawiec, D.; Wen, L.; et al. Caffeine protects against experimental acute pancreatitis by inhibition of inositol 1,4,5-trisphosphate receptor-mediated Ca2+ release. Gut 2017, 66, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Maeda, N.; Niinobe, M.; Mikoshiba, K. A cerebellar Purkinje cell marker P400 protein is an inositol 1,4,5-trisphosphate (InsP3) receptor protein. Purification and characterization of InsP3 receptor complex. EMBO J. 1990, 9, 61–67. [Google Scholar] [CrossRef]
- Hisatsune, C.; Mikoshiba, K. IP3 receptor mutations and brain diseases in human and rodents. J. Neurochem. 2017, 141, 790–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Leemput, J.; Chandran, J.; Knight, M.A.; Holtzclaw, L.A.; Scholz, S.; Cookson, M.R.; Houlden, H.; Gwinn-Hardy, K.; Fung, H.C.; Lin, X.; et al. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 2007, 3, e108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marelli, C.; van de Leemput, J.; Johnson, J.O.; Tison, F.; Thauvin-Robinet, C.; Picard, F.; Tranchant, C.; Hernandez, D.G.; Huttin, B.; Boulliat, J.; et al. SCA15 due to large ITPR1 deletions in a cohort of 333 white families with dominant ataxia. Arch. Neurol. 2011, 68, 637–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, M.J.; Sweeney, M.G.; Li, A.; Treacy, C.; Chandrashekar, H.S.; Giunti, P.; Goold, R.G.; Davis, M.B.; Houlden, H.; Tabrizi, S.J. An ITPR1 gene deletion causes spinocerebellar ataxia 15/16: A genetic, clinical and radiological description. Mov. Disord. 2010, 25, 2176–2182. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Shiga, A.; Nozaki, H.; Mitsui, J.; Takahashi, Y.; Ishiguro, H.; Yomono, H.; Kurisaki, H.; Goto, J.; Ikeuchi, T.; et al. Total deletion and a missense mutation of ITPR1 in Japanese SCA15 families. Neurology 2008, 71, 547–551. [Google Scholar] [CrossRef]
- Ganesamoorthy, D.; Bruno, D.L.; Schoumans, J.; Storey, E.; Delatycki, M.B.; Zhu, D.; Wei, M.K.; Nicholson, G.A.; McKinlay Gardner, R.J.; Slater, H.R. Development of a multiplex ligation-dependent probe amplification assay for diagnosis and estimation of the frequency of spinocerebellar ataxia type 15. Clin. Chem. 2009, 55, 1415–1418. [Google Scholar] [CrossRef] [Green Version]
- Ando, H.; Hirose, M.; Mikoshiba, K. Aberrant IP3 receptor activities revealed by comprehensive analysis of pathological mutations causing spinocerebellar ataxia 29. Proc. Natl. Acad. Sci. USA 2018, 115, 12259–12264. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Chardon, J.W.; Carter, M.T.; Friend, K.L.; Dudding, T.E.; Schwartzentruber, J.; Zou, R.; Schofield, P.W.; Douglas, S.; Bulman, D.E.; et al. Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet. J. Rare Dis. 2012, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Gonzaga-Jauregui, C.; Harel, T.; Gambin, T.; Kousi, M.; Griffin, L.B.; Francescatto, L.; Ozes, B.; Karaca, E.; Jhangiani, S.N.; Bainbridge, M.N.; et al. Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep. 2015, 12, 1169–1183. [Google Scholar] [CrossRef] [Green Version]
- Zambonin, J.L.; Bellomo, A.; Ben-Pazi, H.; Everman, D.B.; Frazer, L.M.; Geraghty, M.T.; Harper, A.D.; Jones, J.R.; Kamien, B.; Kernohan, K.; et al. Spinocerebellar ataxia type 29 due to mutations in ITPR1: A case series and review of this emerging congenital ataxia. Orphanet J. Rare. Dis. 2017, 12, 121. [Google Scholar] [CrossRef]
- Casey, J.P.; Hirouchi, T.; Hisatsune, C.; Lynch, B.; Murphy, R.; Dunne, A.M.; Miyamoto, A.; Ennis, S.; van der Spek, N.; O’Hici, B.; et al. A novel gain-of-function mutation in the ITPR1 suppressor domain causes spinocerebellar ataxia with altered Ca(2+) signal patterns. J. Neurol. 2017, 264, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hao, Y.; Yu, P.; Cao, Z.; Zhang, J.; Zhang, X.; Chen, Y.; Zhang, H.; Gu, W. Identification of a Splicing Mutation in ITPR1 via WES in a Chinese Early-Onset Spinocerebellar Ataxia Family. Cerebellum 2018, 17, 294–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klar, J.; Ali, Z.; Farooq, M.; Khan, K.; Wikstrom, J.; Iqbal, M.; Zulfiqar, S.; Faryal, S.; Baig, S.M.; Dahl, N. A missense variant in ITPR1 provides evidence for autosomal recessive SCA29 with asymptomatic cerebellar hypoplasia in carriers. Eur J. Hum. Genet. 2017, 25, 848–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, M.; Ohba, C.; Iai, M.; Hirabayashi, S.; Osaka, H.; Hiraide, T.; Saitsu, H.; Matsumoto, N. Sporadic infantile-onset spinocerebellar ataxia caused by missense mutations of the inositol 1,4,5-triphosphate receptor type 1 gene. J. Neurol 2015, 262, 1278–1284. [Google Scholar] [CrossRef]
- Fogel, B.L.; Lee, H.; Deignan, J.L.; Strom, S.P.; Kantarci, S.; Wang, X.; Quintero-Rivera, F.; Vilain, E.; Grody, W.W.; Perlman, S.; et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. 2014, 71, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Barresi, S.; Niceta, M.; Alfieri, P.; Brankovic, V.; Piccini, G.; Bruselles, A.; Barone, M.R.; Cusmai, R.; Tartaglia, M.; Bertini, E.; et al. Mutations in the IRBIT domain of ITPR1 are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Clin. Genet. 2017, 91, 86–91. [Google Scholar] [CrossRef]
- Parolin Schnekenberg, R.; Perkins, E.M.; Miller, J.W.; Davies, W.I.; D’Adamo, M.C.; Pessia, M.; Fawcett, K.A.; Sims, D.; Gillard, E.; Hudspith, K.; et al. De novo point mutations in patients diagnosed with ataxic cerebral palsy. Brain 2015, 138, 1817–1832. [Google Scholar] [CrossRef] [Green Version]
- Valencia, C.A.; Husami, A.; Holle, J.; Johnson, J.A.; Qian, Y.; Mathur, A.; Wei, C.; Indugula, S.R.; Zou, F.; Meng, H.; et al. Clinical Impact and Cost-Effectiveness of Whole Exome Sequencing as a Diagnostic Tool: A Pediatric Center’s Experience. Front. Pediatr. 2015, 3, 67. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, C.T.; Liu, Y.T.; Liao, Y.C.; Hsu, T.Y.; Lee, Y.C.; Soong, B.W. Mutational analysis of ITPR1 in a Taiwanese cohort with cerebellar ataxias. PLoS ONE 2017, 12, e0187503. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, S.; Uehara, D.T.; Tanimoto, K.; Mizuno, S.; Chinen, Y.; Fukumura, S.; Takanashi, J.I.; Osaka, H.; Okamoto, N.; Inazawa, J. Comprehensive investigation of CASK mutations and other genetic etiologies in 41 patients with intellectual disability and microcephaly with pontine and cerebellar hypoplasia (MICPCH). PLoS ONE 2017, 12, e0181791. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, T.; Barth, P.; Reneman, L.; Appelhof, B.; Baas, F.; Poll-The, B.T. A de novo missense mutation in the inositol 1,4,5-triphosphate receptor type 1 gene causing severe pontine and cerebellar hypoplasia: Expanding the phenotype of ITPR1-related spinocerebellar ataxia’s. Am. J. Med. Genet. A 2017, 173, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, T.S.; Tu, H.; Nelson, O.; Pook, M.; Hammer, R.; Nukina, N.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci. 2008, 28, 12713–12724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Tang, T.S.; Tu, H.; Nelson, O.; Herndon, E.; Huynh, D.P.; Pulst, S.M.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J. Neurosci. 2009, 29, 9148–9162. [Google Scholar] [CrossRef] [PubMed]
- Kasumu, A.W.; Hougaard, C.; Rode, F.; Jacobsen, T.A.; Sabatier, J.M.; Eriksen, B.L.; Strobaek, D.; Liang, X.; Egorova, P.; Vorontsova, D.; et al. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem. Biol. 2012, 19, 1340–1353. [Google Scholar] [CrossRef] [Green Version]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P. Jr. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Tang, T.S.; Tu, H.; Chan, E.Y.; Maximov, A.; Wang, Z.; Wellington, C.L.; Hayden, M.R.; Bezprozvanny, I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 2003, 39, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.S.; Tu, H.; Orban, P.C.; Chan, E.Y.; Hayden, M.R.; Bezprozvanny, I. HAP1 facilitates effects of mutant huntingtin on inositol 1,4,5-trisphosphate-induced Ca release in primary culture of striatal medium spiny neurons. Eur. J. Neurosci. 2004, 20, 1779–1787. [Google Scholar] [CrossRef]
- Tang, T.S.; Guo, C.; Wang, H.; Chen, X.; Bezprozvanny, I. Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington’s disease mouse model. J. Neurosci. 2009, 29, 1257–1266. [Google Scholar] [CrossRef]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef] [Green Version]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef] [Green Version]
- Hirashima, N.; Etcheberrigaray, R.; Bergamaschi, S.; Racchi, M.; Battaini, F.; Binetti, G.; Govoni, S.; Alkon, D.L. Calcium responses in human fibroblasts: A diagnostic molecular profile for Alzheimer’s disease. Neurobiol. Aging 1996, 17, 549–555. [Google Scholar] [CrossRef]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C. Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-beta peptide. J. Neurosci. Res. 2004, 76, 872–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, F.D. Aniridia, Cerebellar Ataxia, and Oligophrenia in Siblings. Arch. Ophthalmol. 1965, 73, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.; Alzayady, K.J.; Burglen, L.; Bremond-Gignac, D.; Marchesin, V.; Roche, O.; Rio, M.; Funalot, B.; Calmon, R.; Durr, A.; et al. Recessive and Dominant De Novo ITPR1 Mutations Cause Gillespie Syndrome. Am. J. Hum. Genet. 2016, 98, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Dentici, M.L.; Barresi, S.; Nardella, M.; Bellacchio, E.; Alfieri, P.; Bruselles, A.; Pantaleoni, F.; Danieli, A.; Iarossi, G.; Cappa, M.; et al. Identification of novel and hotspot mutations in the channel domain of ITPR1 in two patients with Gillespie syndrome. Gene 2017, 628, 141–145. [Google Scholar] [CrossRef]
- McEntagart, M.; Williamson, K.A.; Rainger, J.K.; Wheeler, A.; Seawright, A.; De Baere, E.; Verdin, H.; Bergendahl, L.T.; Quigley, A.; Rainger, J.; et al. A Restricted Repertoire of De Novo Mutations in ITPR1 Cause Gillespie Syndrome with Evidence for Dominant-Negative Effect. Am. J. Hum. Genet. 2016, 98, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Schmunk, G.; Gargus, J.J. Channelopathy pathogenesis in autism spectrum disorders. Front. Genet. 2013, 4, 222. [Google Scholar] [CrossRef] [Green Version]
- Cross-Disorder Group of the Psychiatric Genomics, C. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 2013, 381, 1371–1379. [Google Scholar]
- Schmunk, G.; Boubion, B.J.; Smith, I.F.; Parker, I.; Gargus, J.J. Shared functional defect in IP(3)R-mediated calcium signaling in diverse monogenic autism syndromes. Transl. Psychiatry 2015, 5, e643. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.J.; Hashii, M.; Munesue, T.; Hayashi, K.; Yagi, K.; Yamagishi, M.; Higashida, H.; Yokoyama, S. Non-synonymous single-nucleotide variations of the human oxytocin receptor gene and autism spectrum disorders: A case-control study in a Japanese population and functional analysis. Mol. Autism 2013, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Van Den Bosch, L.; Verhoeven, K.; De Smedt, H.; Wuytack, F.; Missiaen, L.; Robberecht, W. Calcium handling proteins in isolated spinal motoneurons. Life Sci. 1999, 65, 1597–1606. [Google Scholar] [CrossRef]
- van Es, M.A.; Van Vught, P.W.; Blauw, H.M.; Franke, L.; Saris, C.G.; Andersen, P.M.; Van Den Bosch, L.; de Jong, S.W.; van ’t Slot, R.; Birve, A.; et al. ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: A genome-wide association study. Lancet Neurol. 2007, 6, 869–877. [Google Scholar] [CrossRef]
- Kim, S.H.; Zhan, L.; Hanson, K.A.; Tibbetts, R.S. High-content RNAi screening identifies the Type 1 inositol triphosphate receptor as a modifier of TDP-43 localization and neurotoxicity. Hum. Mol. Genet. 2012, 21, 4845–4856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futatsugi, A.; Nakamura, T.; Yamada, M.K.; Ebisui, E.; Nakamura, K.; Uchida, K.; Kitaguchi, T.; Takahashi-Iwanaga, H.; Noda, T.; Aruga, J.; et al. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 2005, 309, 2232–2234. [Google Scholar] [CrossRef]
- Miyachi, K.; Iwai, M.; Asada, K.; Saito, I.; Hankins, R.; Mikoshiba, K. Inositol 1,4,5-trisphosphate receptors are autoantibody target antigens in patients with Sjogren’s syndrome and other systemic rheumatic diseases. Mod. Rheumatol. 2007, 17, 137–143. [Google Scholar] [CrossRef]
- Vivino, F.B. Sjogren’s syndrome: Clinical aspects. Clin. Immunol. 2017, 182, 48–54. [Google Scholar] [CrossRef]
- Teos, L.Y.; Zhang, Y.; Cotrim, A.P.; Swaim, W.; Won, J.H.; Ambrus, J.; Shen, L.; Bebris, L.; Grisius, M.; Jang, S.I.; et al. IP3R deficit underlies loss of salivary fluid secretion in Sjogren’s Syndrome. Sci. Rep. 2015, 5, 13953. [Google Scholar] [CrossRef] [Green Version]
- Klar, J.; Hisatsune, C.; Baig, S.M.; Tariq, M.; Johansson, A.C.; Rasool, M.; Malik, N.A.; Ameur, A.; Sugiura, K.; Feuk, L.; et al. Abolished InsP3R2 function inhibits sweat secretion in both humans and mice. J. Clin. Invest. 2014, 124, 4773–4780. [Google Scholar] [CrossRef]
- Sneyers, F.; Rosa, N.; Bultynck, G. Type 3 IP3 receptors driving oncogenesis. Cell Calcium 2020, 86, 102141. [Google Scholar] [CrossRef]
- Hedberg, M.L.; Goh, G.; Chiosea, S.I.; Bauman, J.E.; Freilino, M.L.; Zeng, Y.; Wang, L.; Diergaarde, B.B.; Gooding, W.E.; Lui, V.W.; et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J. Clin. Invest. 2016, 126, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Rezuchova, I.; Hudecova, S.; Soltysova, A.; Matuskova, M.; Durinikova, E.; Chovancova, B.; Zuzcak, M.; Cihova, M.; Burikova, M.; Penesova, A.; et al. Type 3 inositol 1,4,5-trisphosphate receptor has antiapoptotic and proliferative role in cancer cells. Cell Death Dis. 2019, 10, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueasilamongkol, P.; Khamphaya, T.; Guerra, M.T.; Rodrigues, M.A.; Gomes, D.A.; Kong, Y.; Wei, W.; Jain, D.; Trampert, D.C.; Ananthanarayanan, M.; et al. Weerachayaphorn, J. Type 3 Inositol 1,4,5-Trisphosphate Receptor Is Increased and Enhances Malignant Properties in Cholangiocarcinoma. Hepatology 2020, 71, 583–599. [Google Scholar] [CrossRef] [PubMed]
- Shibao, K.; Fiedler, M.J.; Nagata, J.; Minagawa, N.; Hirata, K.; Nakayama, Y.; Iwakiri, Y.; Nathanson, M.H.; Yamaguchi, K. The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 2010, 48, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, A.; Rabionet, R.; Espinet, B.; Zapata, L.; Puiggros, A.; Melero, C.; Puig, A.; Sarria-Trujillo, Y.; Ossowski, S.; Garcia-Muret, M.P.; et al. Identification of Gene Mutations and Fusion Genes in Patients with Sezary Syndrome. J. Invest. Derm. 2016, 136, 1490–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleftherohorinou, H.; Hoggart, C.J.; Wright, V.J.; Levin, M.; Coin, L.J. Pathway-driven gene stability selection of two rheumatoid arthritis GWAS identifies and validates new susceptibility genes in receptor mediated signalling pathways. Hum. Mol. Genet. 2011, 20, 3494–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torkamani, A.; Topol, E.J.; Schork, N.J. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics 2008, 92, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Nakabayashi, K.; Tajima, A.; Yamamoto, K.; Takahashi, A.; Hata, K.; Takashima, Y.; Koyanagi, M.; Nakaoka, H.; Akamizu, T.; Ishikawa, N.; et al. Identification of independent risk loci for Graves’ disease within the MHC in the Japanese population. J. Hum. Genet. 2011, 56, 772–778. [Google Scholar] [CrossRef]
- Ferreira, M.A.; Vonk, J.M.; Baurecht, H.; Marenholz, I.; Tian, C.; Hoffman, J.D.; Helmer, Q.; Tillander, A.; Ullemar, V.; van Dongen, J.; et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat. Genet. 2017, 49, 1752–1757. [Google Scholar] [CrossRef]
- Kichaev, G.; Bhatia, G.; Loh, P.R.; Gazal, S.; Burch, K.; Freund, M.K.; Schoech, A.; Pasaniuc, B.; Price, A.L. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am. J. Hum. Genet. 2019, 104, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Wilk, J.B.; Shrine, N.R.; Loehr, L.R.; Zhao, J.H.; Manichaikul, A.; Lopez, L.M.; Smith, A.V.; Heckbert, S.R.; Smolonska, J.; Tang, W.; et al. Genome-wide association studies identify CHRNA5/3 and HTR4 in the development of airflow obstruction. Am. J. Respir. Crit. Care Med. 2012, 186, 622–632. [Google Scholar] [CrossRef] [Green Version]
- Reddy, M.V.; Wang, H.; Liu, S.; Bode, B.; Reed, J.C.; Steed, R.D.; Anderson, S.W.; Steed, L.; Hopkins, D.; She, J.X. Association between type 1 diabetes and GWAS SNPs in the southeast US Caucasian population. Genes Immun. 2011, 12, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Roach, J.C.; Deutsch, K.; Li, S.; Siegel, A.F.; Bekris, L.M.; Einhaus, D.C.; Sheridan, C.M.; Glusman, G.; Hood, L.; Lernmark, A.; et al. Swedish Childhood Diabetes Study, G.; Diabetes Incidence in Sweden Study, G. Genetic mapping at 3-kilobase resolution reveals inositol 1,4,5-triphosphate receptor 3 as a risk factor for type 1 diabetes in Sweden. Am. J. Hum. Genet. 2006, 79, 614–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, T.; Iida, A.; Otsubo, S.; Kamatani, Y.; Usami, M.; Takei, T.; Uchida, K.; Tsuchiya, K.; Saito, S.; Ohnisi, Y.; et al. A functional SNP in the NKX2.5-binding site of ITPR3 promoter is associated with susceptibility to systemic lupus erythematosus in Japanese population. J. Hum. Genet. 2008, 53, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Cotsapas, C.; Speliotes, E.K.; Hatoum, I.J.; Greenawalt, D.M.; Dobrin, R.; Lum, P.Y.; Suver, C.; Chudin, E.; Kemp, D.; Reitman, M.; et al. Consortium, G. Common body mass index-associated variants confer risk of extreme obesity. Hum. Mol. Genet. 2009, 18, 3502–3507. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.K.; Sedor, J.R.; Freedman, B.I.; Kao, W.H.; Kretzler, M.; Keller, B.J.; Abboud, H.E.; Adler, S.G.; Best, L.G.; Bowden, D.W.; et al. Diabetes, Genome-Wide Association and Trans-ethnic Meta-Analysis for Advanced Diabetic Kidney Disease: Family Investigation of Nephropathy and Diabetes (FIND). PLoS Genet. 2015, 11, e1005352. [Google Scholar] [CrossRef] [PubMed]
- Comuzzie, A.G.; Cole, S.A.; Laston, S.L.; Voruganti, V.S.; Haack, K.; Gibbs, R.A.; Butte, N.F. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS ONE 2012, 7, e51954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Wen, Y.; Guo, X.; Zhang, Y.; Wang, X.; Yang, T.; Shen, H.; Chen, X.; Tian, Q.; Deng, H.W. Genome-wide association study identifies ITPR2 as a susceptibility gene for Kashin-Beck disease in Han Chinese. Arthritis Rheumatol. 2015, 67, 176–181. [Google Scholar] [CrossRef]
- Wang, S.J.; Guo, X.; Zuo, H.; Zhang, Y.G.; Xu, P.; Ping, Z.G.; Zhang, Z.; Geng, D. Chondrocyte apoptosis and expression of Bcl-2, Bax, Fas, and iNOS in articular cartilage in patients with Kashin-Beck disease. J. Rheumatol. 2006, 33, 615–619. [Google Scholar]
- Mirza, N.; Appleton, R.; Burn, S.; Carr, D.; Crooks, D.; du Plessis, D.; Duncan, R.; Farah, J.O.; Josan, V.; Miyajima, F.; et al. Identifying the biological pathways underlying human focal epilepsy: From complexity to coherence to centrality. Hum. Mol. Genet. 2015, 24, 4306–4316. [Google Scholar] [CrossRef]
- Nagarkatti, N.; Deshpande, L.S.; DeLorenzo, R.J. Levetiracetam inhibits both ryanodine and IP3 receptor activated calcium induced calcium release in hippocampal neurons in culture. Neurosci. Lett. 2008, 436, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Meder, B.; Haas, J.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Frese, K.; Lai, A.; Nietsch, R.; Scheiner, C.; Mester, S.; Bordalo, D.M.; et al. Epigenome-Wide Association Study Identifies Cardiac Gene Patterning and a Novel Class of Biomarkers for Heart Failure. Circulation 2017, 136, 1528–1544. [Google Scholar] [CrossRef] [PubMed]
- Consortium, C.A.D.; Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; Ingelsson, E.; Saleheen, D.; Erdmann, J.; et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013, 45, 25–33. [Google Scholar]
- Huang, Y.C.; Lin, Y.J.; Chang, J.S.; Chen, S.Y.; Wan, L.; Sheu, J.J.; Lai, C.H.; Lin, C.W.; Liu, S.P.; Chen, C.P.; et al. Single nucleotide polymorphism rs2229634 in the ITPR3 gene is associated with the risk of developing coronary artery aneurysm in children with Kawasaki disease. Int J. Immunogenet. 2010, 37, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Bijnens, J.; Missiaen, L.; Bultynck, G.; Parys, J.B. A critical appraisal of the role of intracellular Ca(2+)-signaling pathways in Kawasaki disease. Cell Calcium 2018, 71, 95–103. [Google Scholar] [CrossRef]
- Nakazawa, M.; Uchida, K.; Aramaki, M.; Kodo, K.; Yamagishi, C.; Takahashi, T.; Mikoshiba, K.; Yamagishi, H. Inositol 1,4,5-trisphosphate receptors are essential for the development of the second heart field. J. Mol. Cell Cardiol. 2011, 51, 58–66. [Google Scholar] [CrossRef]
- Uchida, K.; Aramaki, M.; Nakazawa, M.; Yamagishi, C.; Makino, S.; Fukuda, K.; Nakamura, T.; Takahashi, T.; Mikoshiba, K.; Yamagishi, H. Gene knock-outs of inositol 1,4,5-trisphosphate receptors types 1 and 2 result in perturbation of cardiogenesis. PLoS ONE 2010, 5, e12500. [Google Scholar] [CrossRef] [Green Version]
- Uchida, K.; Nakazawa, M.; Yamagishi, C.; Mikoshiba, K.; Yamagishi, H. Type 1 and 3 inositol trisphosphate receptors are required for extra-embryonic vascular development. Dev. Biol 2016, 418, 89–97. [Google Scholar] [CrossRef]
- Miyazaki, S.; Yuzaki, M.; Nakada, K.; Shirakawa, H.; Nakanishi, S.; Nakade, S.; Mikoshiba, K. Block of Ca2+ wave and Ca2+ oscillation by antibody to the inositol 1,4,5-trisphosphate receptor in fertilized hamster eggs. Science 1992, 257, 251–255. [Google Scholar] [CrossRef]
- Saneyoshi, T.; Kume, S.; Amasaki, Y.; Mikoshiba, K. The Wnt/calcium pathway activates NF-AT and promotes ventral cell fate in Xenopus embryos. Nature 2002, 417, 295–299. [Google Scholar] [CrossRef]
- Parmar, P.G.; Taal, H.R.; Timpson, N.J.; Thiering, E.; Lehtimaki, T.; Marinelli, M.; Lind, P.A.; Howe, L.D.; Verwoert, G.; Aalto, V.; et al. International Genome-Wide Association Study Consortium Identifies Novel Loci Associated With Blood Pressure in Children and Adolescents. Circ. Cardiovasc. Genet. 2016, 9, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Michailidou, K.; Beesley, J.; Lindstrom, S.; Canisius, S.; Dennis, J.; Lush, M.J.; Maranian, M.J.; Bolla, M.K.; Wang, Q.; Shah, M.; et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015, 47, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michailidou, K.; Lindstrom, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemacon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Kim, J.; Kim, S.W.; Park, S.K.; Ahn, S.H.; Lee, M.H.; Suh, Y.J.; Noh, D.Y.; Son, B.H.; Cho, Y.U.; et al. BRCA1/2-negative, high-risk breast cancers (BRCAX) for Asian women: Genetic susceptibility loci and their potential impacts. Sci Rep. 2018, 8, 15263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakura, C.; Hagiwara, A.; Fukuda, K.; Shimomura, K.; Takagi, T.; Kin, S.; Nakase, Y.; Fujiyama, J.; Mikoshiba, K.; Okazaki, Y.; et al. Possible involvement of inositol 1,4,5-trisphosphate receptor type 3 (IP3R3) in the peritoneal dissemination of gastric cancers. Anticancer Res. 2003, 23, 3691–3697. [Google Scholar] [PubMed]
- Yang, Y.C.; Chang, T.Y.; Chen, T.C.; Lin, W.S.; Chang, S.C.; Lee, Y.J. ITPR3 gene haplotype is associated with cervical squamous cell carcinoma risk in Taiwanese women. Oncotarget 2017, 8, 10085–10090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackshaw, S.; Sawa, A.; Sharp, A.H.; Ross, C.A.; Snyder, S.H.; Khan, A.A. Type 3 inositol 1,4,5-trisphosphate receptor modulates cell death. FASEB J. 2000, 14, 1375–1379. [Google Scholar]
- Mendes, C.C.; Gomes, D.A.; Thompson, M.; Souto, N.C.; Goes, T.S.; Goes, A.M.; Rodrigues, M.A.; Gomez, M.V.; Nathanson, M.H.; Leite, M.F. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem. 2005, 280, 40892–40900. [Google Scholar] [CrossRef] [Green Version]
- Bartok, A.; Weaver, D.; Golenar, T.; Nichtova, Z.; Katona, M.; Bansaghi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 3726. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Vegas, A.R.; Cordova, A.; Valladares, D.; Llanos, P.; Hidalgo, C.; Gherardi, G.; De Stefani, D.; Mammucari, C.; Rizzuto, R.; Contreras-Ferrat, A.; et al. Mitochondrial Calcium Increase Induced by RyR1 and IP3R Channel Activation After Membrane Depolarization Regulates Skeletal Muscle Metabolism. Front. Physiol. 2018, 9, 791. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Jana, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernandez, E.; Sassano, M.L.; et al. Non-canonical function of IRE1alpha determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nature cell biology 2019, 21, 755–767. [Google Scholar] [CrossRef]
- Cardenas, C.; Muller, M.; McNeal, A.; Lovy, A.; Jana, F.; Bustos, G.; Urra, F.; Smith, N.; Molgo, J.; Diehl, J.A.; et al. Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep. 2016, 14, 2313–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arruda, A.P.; Pers, B.M.; Parlakgul, G.; Guney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straub, S.V.; Giovannucci, D.R.; Yule, D.I. Calcium wave propagation in pancreatic acinar cells: Functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. J. Gen. Physiol. 2000, 116, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiel, C.; Lallet-Daher, H.; Gitenay, D.; Gras, B.; Le Calve, B.; Augert, A.; Ferrand, M.; Prevarskaya, N.; Simonnet, H.; Vindrieux, D.; et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014, 5, 3792. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca(2+) flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [Green Version]
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Marinello, M.; Bononi, A.; Bonora, M.; Giorgi, C.; Rimessi, A.; Pinton, P. Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis. 2012, 3, e304. [Google Scholar] [CrossRef] [Green Version]
- Suman, M.; Sharpe, J.A.; Bentham, R.B.; Kotiadis, V.N.; Menegollo, M.; Pignataro, V.; Molgo, J.; Muntoni, F.; Duchen, M.R.; Pegoraro, E.; et al. Inositol trisphosphate receptor-mediated Ca2+ signalling stimulates mitochondrial function and gene expression in core myopathy patients. Hum. Mol. Genet. 2018, 27, 2367–2382. [Google Scholar] [CrossRef]
- Hohendanner, F.; Maxwell, J.T.; Blatter, L.A. Cytosolic and nuclear calcium signaling in atrial myocytes: IP3-mediated calcium release and the role of mitochondria. Channels (Austin) 2015, 9, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, H.; Nozaki, H.; Onodera, O.; Michikawa, T.; Nishizawa, M.; Mikoshiba, K. Functional characterization of the P1059L mutation in the inositol 1,4,5-trisphosphate receptor type 1 identified in a Japanese SCA15 family. Biochem. Biophys. Res. Commun. 2011, 410, 754–758. [Google Scholar] [CrossRef] [PubMed]


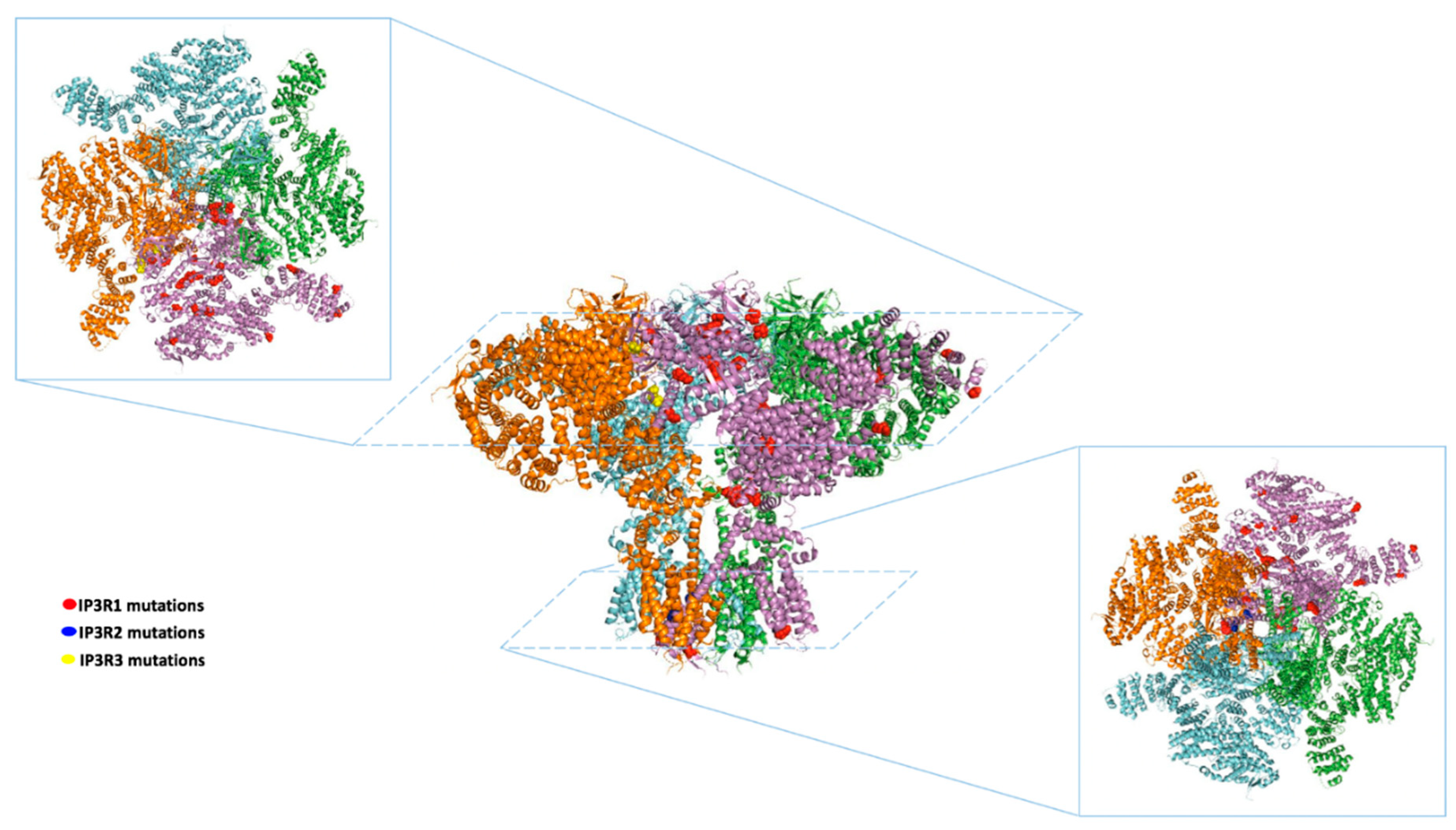{kind=link}
| Tissue | IP3R1 | IP3R2 | IP3R3 |
|---|---|---|---|
| Cerebral cortex | XX | XX | X |
| Cerebellum | XX | X | XXX |
| Hippocampus | XX | ||
| Caudate | XX | X | |
| Thyroid gland | X | X | |
| Parathyroid gland | XXX | ||
| Adrenal gland | XX | X | |
| Nasopharynx | X | XX | |
| Bronchus | XX | XX | |
| Lung | X | X | XX |
| Oral mucosa | X | XX | |
| Salivary gland | XX | ||
| Esophagus | X | XX | |
| Stomach | X | XX | XX |
| Duodenum | XX | XX | |
| Small intestine | XX | XXX | |
| Colon | XX | XX | |
| Rectum | XX | XX | |
| Liver | XX | XX | |
| Gallbladder | XX | X | |
| Pancreas | XX | X | |
| Kidney | X | XXX | X |
| Urinary bladder | X | XX | |
| Testis | X | XX | XXX |
| Epididymis | X | XX | X |
| Seminal vesicle | X | X | X |
| Prostate | X | X | |
| Vagina | XX | ||
| Ovary | X | ||
| Fallopian tube | XX | X | |
| Endometrium | XX | XXX | |
| Cervix, uterine | X | XX | |
| Placenta | X | X | |
| Breast | X | XXX | XX |
| Heart | X | XX | |
| Smooth muscle | XX | ||
| Skeletal muscle | XX | ||
| Soft tissue | |||
| Adipose tissue | XX | ||
| Skin | XX | XX | |
| Appendix | XX | XX | |
| Spleen | X | ||
| Lymph node | X | X | |
| Tonsil | X | X | XXX |
| Bone marrow | X |
| Mutation | IP3R Isoform | Effect on Protein | Disease | Reference |
|---|---|---|---|---|
| 5′ deletion | IP3R1 | Downregulation | SCA15 | [32] |
| 1-48 exons deletion | IP3R1 | Downregulation | SCA15-16 | [33,34] |
| P1059L | IP3R1 | Missense (ND) | SCA15 | [35] |
| P1074L | IP3R1 | Missense (ND) | SCA15 | [35] |
| V494I | IP3R1 | Missense (ND) | SCA15 | [36] |
| V1553M | IP3R1 | Missense (ND) | SCA29 | [38] |
| N602D | IP3R1 | Missense (ND) | SCA29 | [38] |
| G2547A | IP3R1 | Missense (ND) | SCA29 | [39] |
| R269G | IP3R1 | Missense (ND) | SCA29 | [40] |
| K279E | IP3R1 | Missense (ND) | SCA29 | [40] |
| G2506R | IP3R1 | Missense (ND) | SCA29 | [40] |
| I2550T | IP3R1 | Missense (ND) | SCA29 | [40] |
| T1386M | IP3R1 | Missense (ND) | SCA29 | [40] |
| R36C | IP3R1 | Gain-of-function Increase of IP3 binding affinity | SCA29 | [41] |
| c.1207-2A-T | IP3R1 | Splicing variant | SCA29 | [42] |
| L1787P | IP3R1 | Protein-instability* | Autosomal-recessive SCA | [43] |
| T267M | IP3R1 | Missense (ND) | Sporadic infantile-onset-SCA | [44,45] |
| T594I | IP3R1 | Missense (ND) | Sporadic infantile-onset-SCA | [44,45] |
| S277I | IP3R1 | Missense (ND) | Sporadic infantile-onset-SCA | [44,45] |
| T267R | IP3R1 | Missense (ND) | Sporadic infantile-onset-SCA | [44,45] |
| R269W | IP3R1 | Missense (ND) | Congenital-ataxias | [46] |
| R241K | IP3R1 | Missense (ND) | Congenital-ataxias | [46] |
| A280D | IP3R1 | Missense (ND) | Congenital-ataxias | [46] |
| E512K | IP3R1 | Missense (ND) | Congenital-ataxias | [46] |
| S1493D | IP3R1 | Missense (ND) | Ataxic-cerebral-palsy | [47] |
| V2541A | IP3R1 | Missense (ND) | Molecular-unassigned SCA | [48] |
| T2490M | IP3R1 | Missense (ND) | Molecular-unassigned SCA | [48] |
| T2552P | IP3R1 | Missense (ND) | Cerebellar-hypoplasia | [50] |
| I2550N | IP3R1 | Missense (ND) | Cerebellar-hypoplasia | [51] |
| Q1558 | IP3R1 | Truncating-protein, no functional channel | Gillespie syndrome | [64] |
| R728 | IP3R1 | Truncating-protein, no functional channel | Gillespie syndrome | [64] |
| F2553L | IP3R1 | Missense (ND) | Gillespie syndrome | [64] |
| K2563 deletion | IP3R1 | Dysfunctional channel with dominant negative action | Gillespie syndrome | [64] |
| N2543I | IP3R1 | Missense (ND) | Gillespie syndrome | [65] |
| E2061G | IP3R1 | Missense (ND) | Gillespie syndrome | [66] |
| E2061Q | IP3R1 | Missense (ND) | Gillespie syndrome | [66] |
| A95T | IP3R1 | Missense (ND) | Sézary syndrome | [84] |
| S2454F | IP3R1 | Missense (ND) | Sézary syndrome | [84] |
| S2508L | IP3R1 | Missense (ND) | Sézary syndrome | [84] |
| G2498S | IP3R2 | Missense: dysfunctional channel * | Anhidrosis | [78] |
| R64H | IP3R3 | Missense (ND) | HNSCC | [80] |
| R149L | IP3R3 | Missense (ND) | HNSCC | [80] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambardella, J.; Lombardi, A.; Morelli, M.B.; Ferrara, J.; Santulli, G. Inositol 1,4,5-Trisphosphate Receptors in Human Disease: A Comprehensive Update. J. Clin. Med. 2020, 9, 1096. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9041096
Gambardella J, Lombardi A, Morelli MB, Ferrara J, Santulli G. Inositol 1,4,5-Trisphosphate Receptors in Human Disease: A Comprehensive Update. Journal of Clinical Medicine. 2020; 9(4):1096. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9041096
Chicago/Turabian StyleGambardella, Jessica, Angela Lombardi, Marco Bruno Morelli, John Ferrara, and Gaetano Santulli. 2020. "Inositol 1,4,5-Trisphosphate Receptors in Human Disease: A Comprehensive Update" Journal of Clinical Medicine 9, no. 4: 1096. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9041096