
New Functional Bionanocomposites by Combining Hybrid Host-Guest Systems with a Fully Biobased Poly(lactic acid)/Poly(butylene succinate-co-adipate) (PLA/PBSA) Binary Blend

 ,
,  ,
,  ,
,  , ,
, ,  and
and 
Abstract
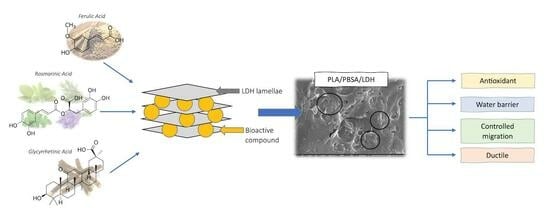
:
1. Introduction
2. Materials and Methods
2.1. Materials
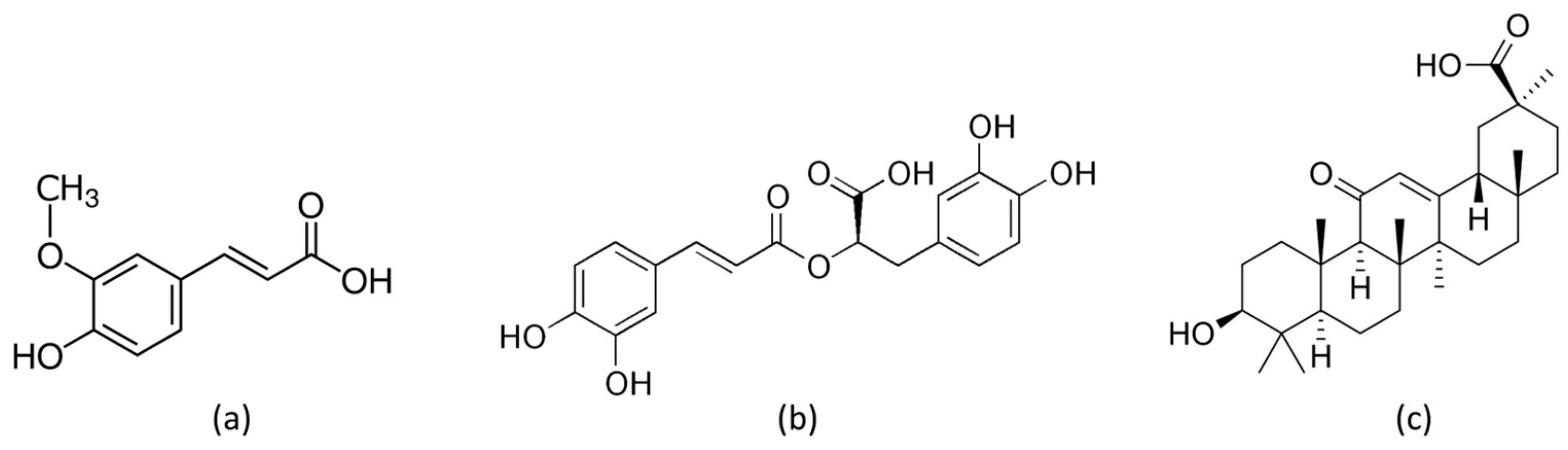
2.2. Preparation of Modified LDHs (LDH-FA, LDH-RA, and LDH-GA)
2.3. Preparation of Polymer Nanocomposites
2.3.1. Melt Compounding in a Batch Mixer
2.3.2. Melt Compounding in a Micro-Compounder
2.4. Characterization
2.5. DPPH Test
2.6. Migration Test
3. Results
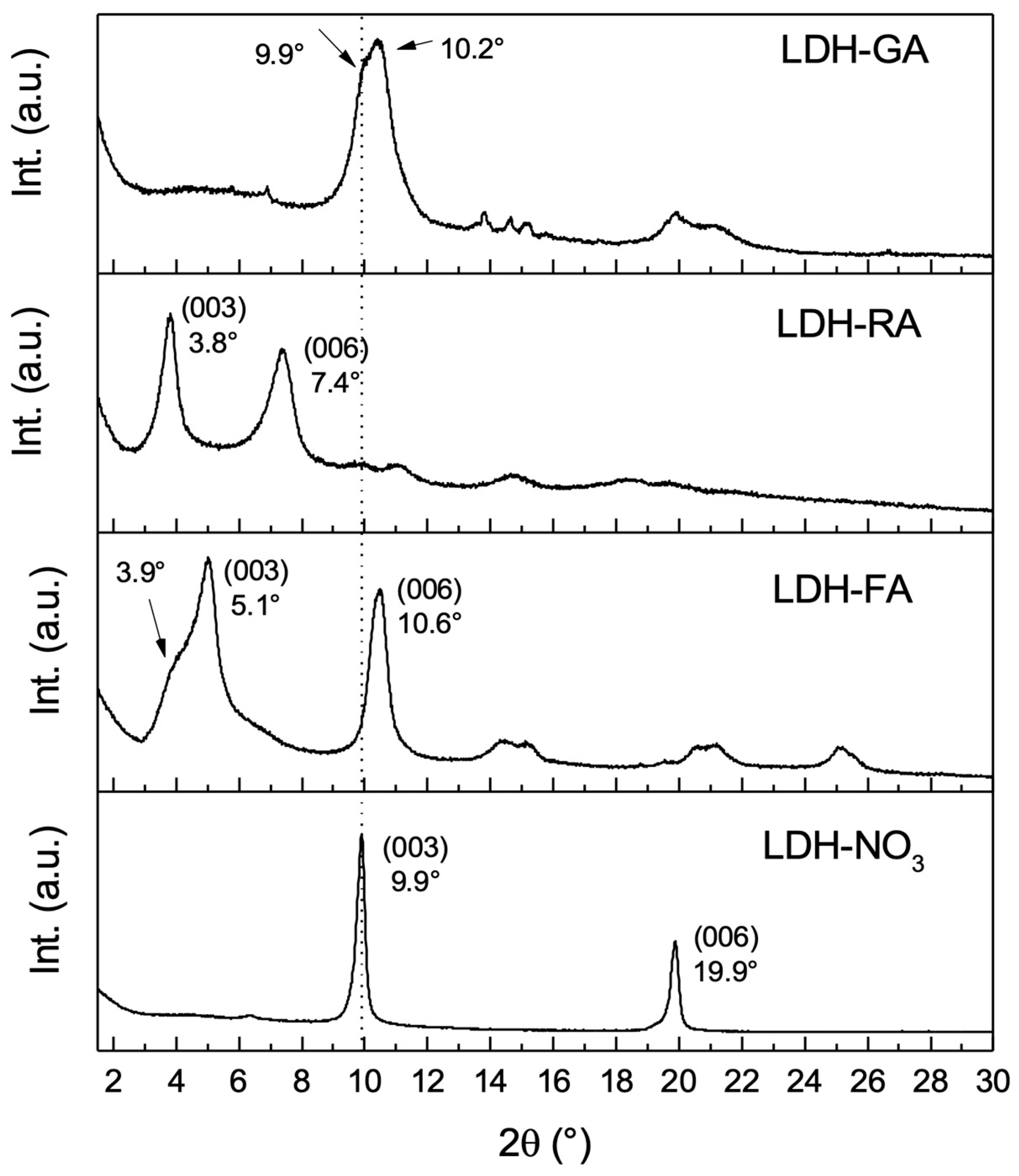
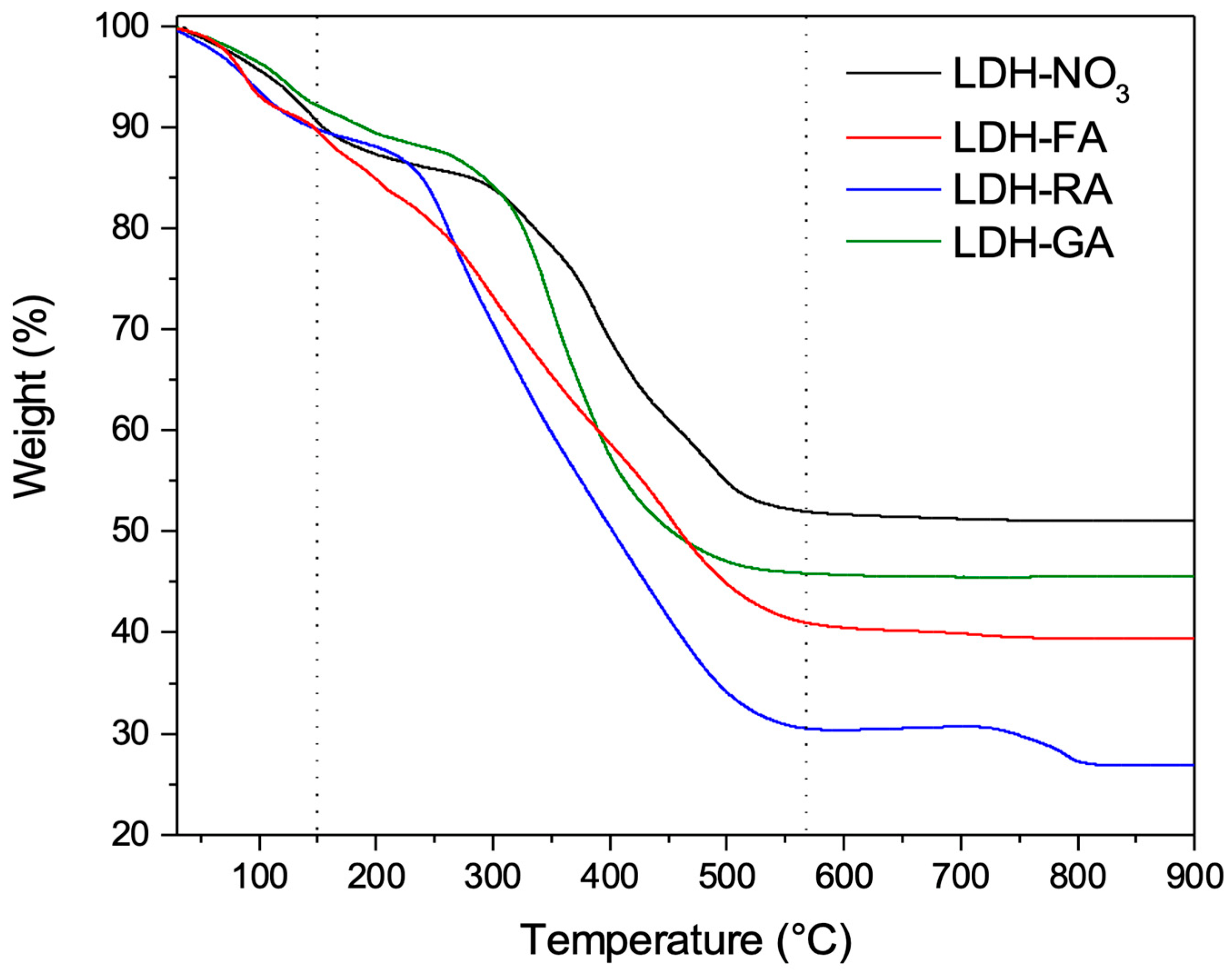
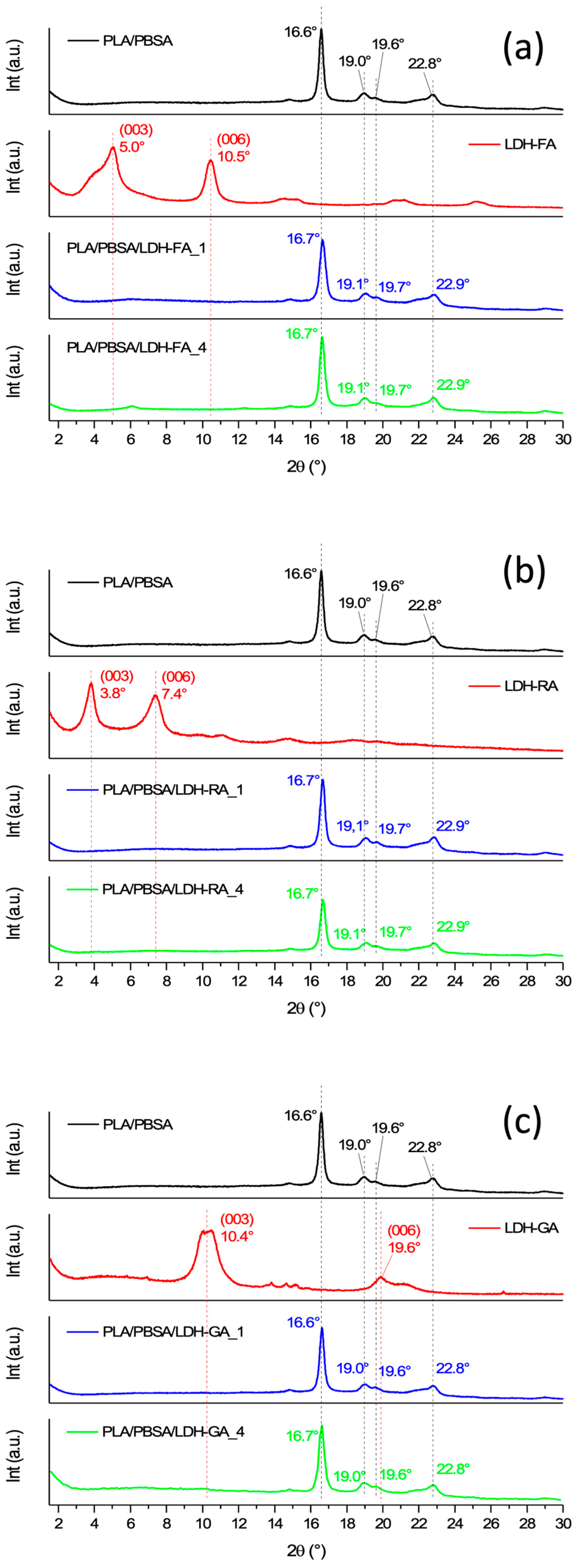
3.1. Characterization of Functional LDHs
3.2. PLA/PBSA/LDH Nanocomposites via Batch Mixing
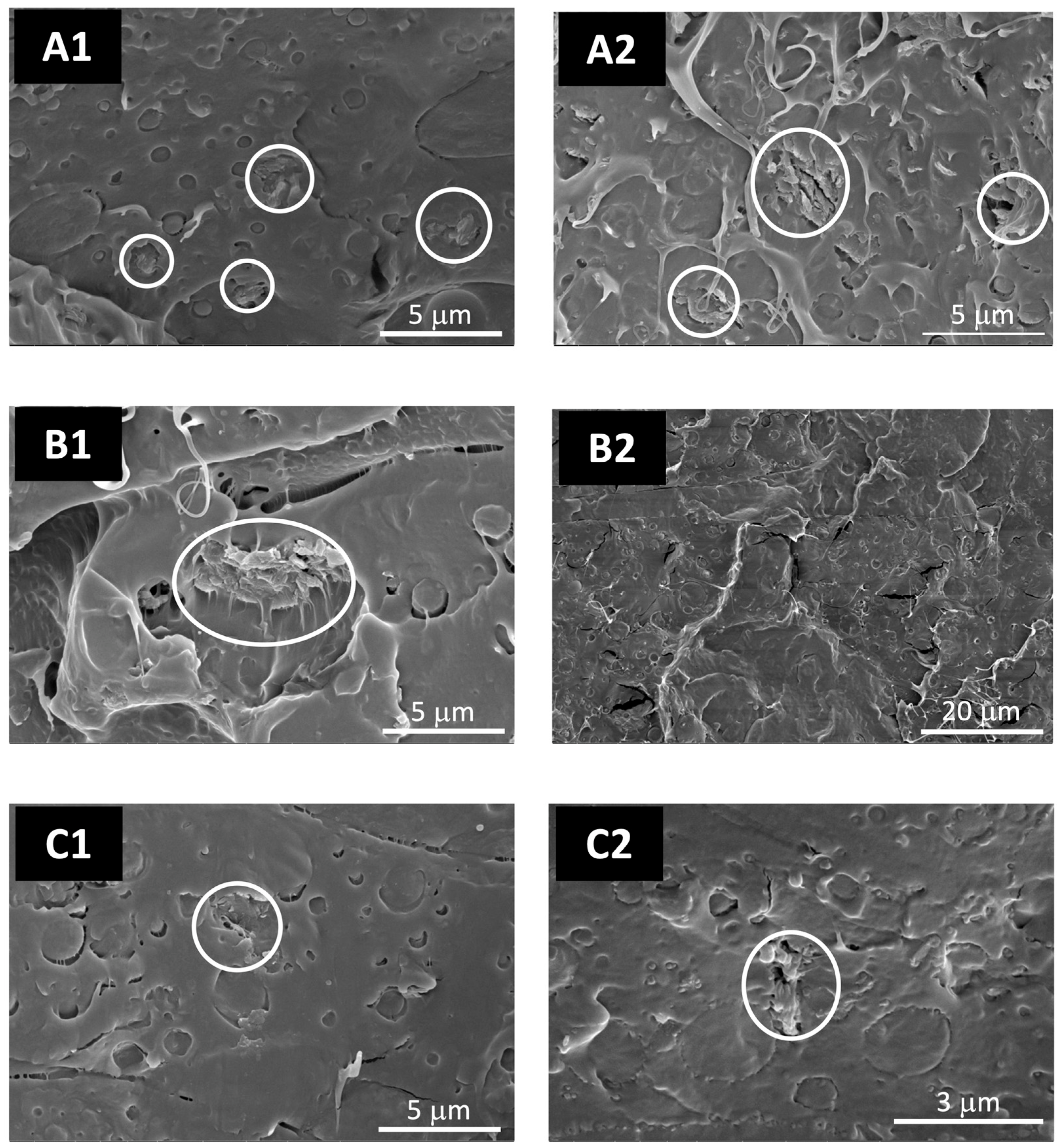
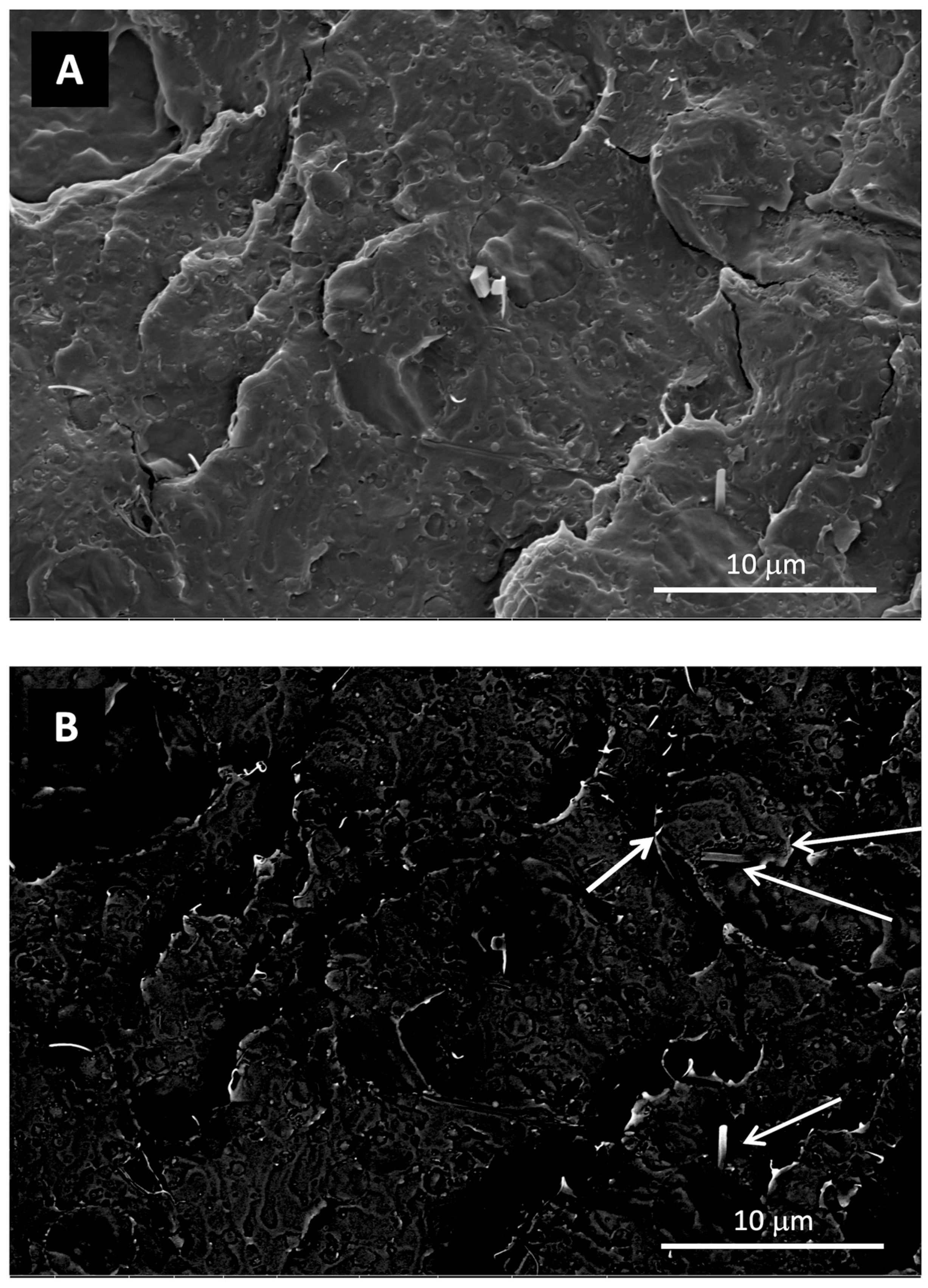
3.2.1. Structure and Morphology
3.2.2. TGA Analysis
3.2.3. SEC Analysis
3.2.4. Antioxidant Properties
3.2.5. Water Vapor Permeability
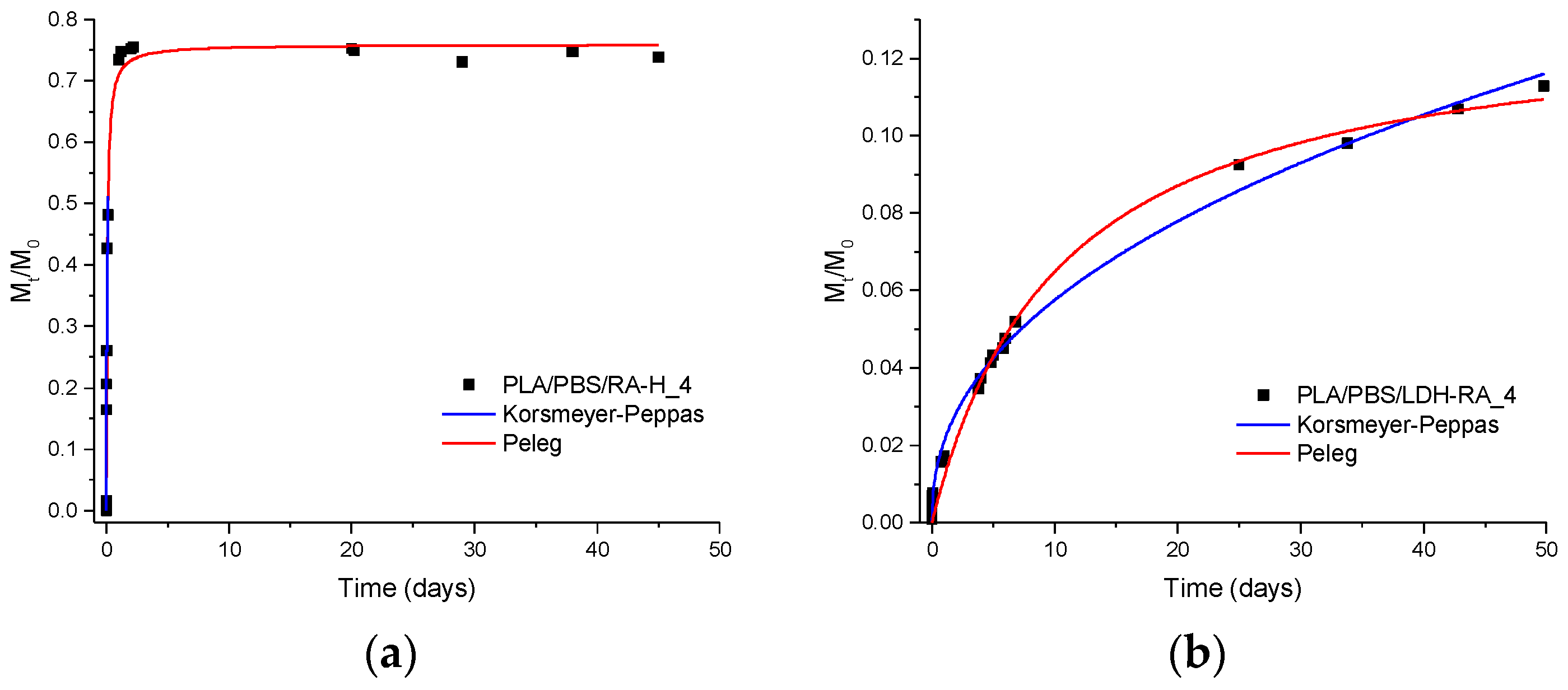
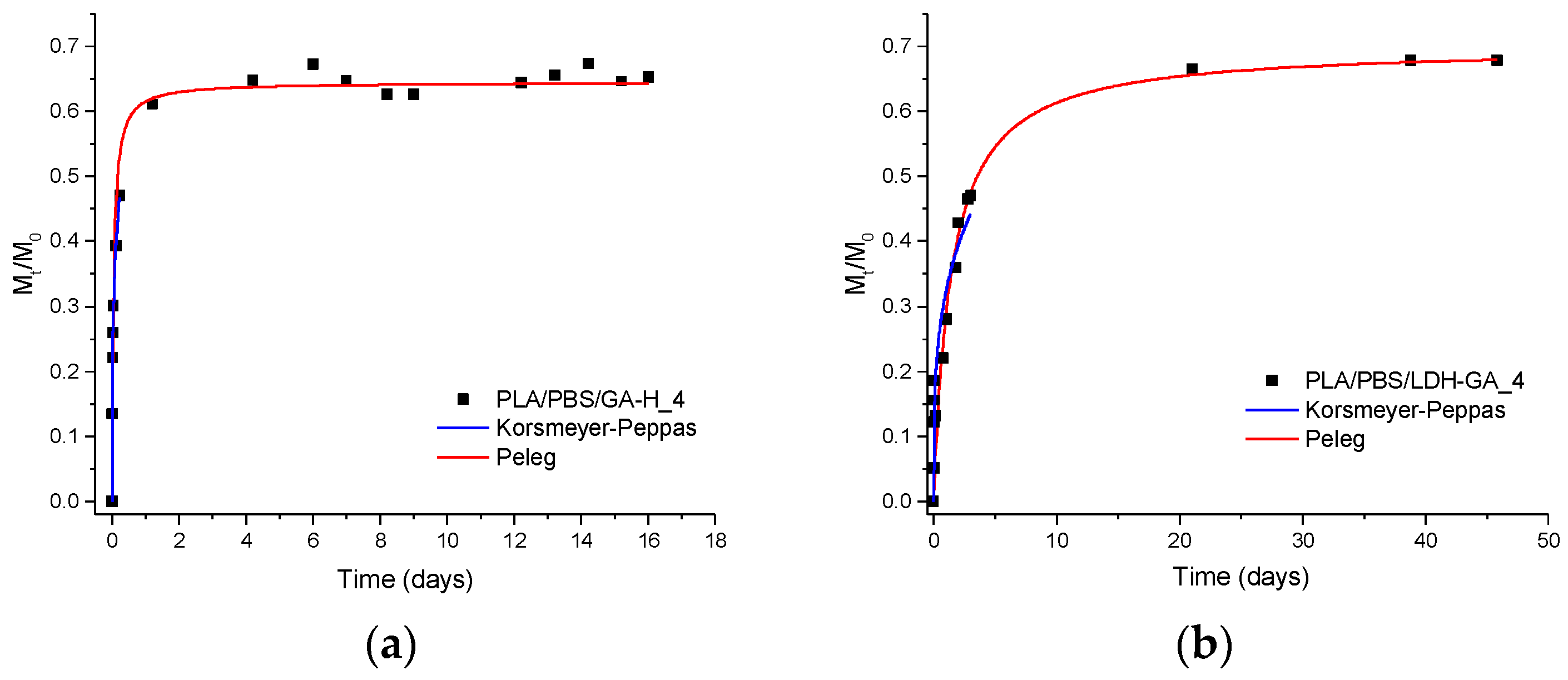
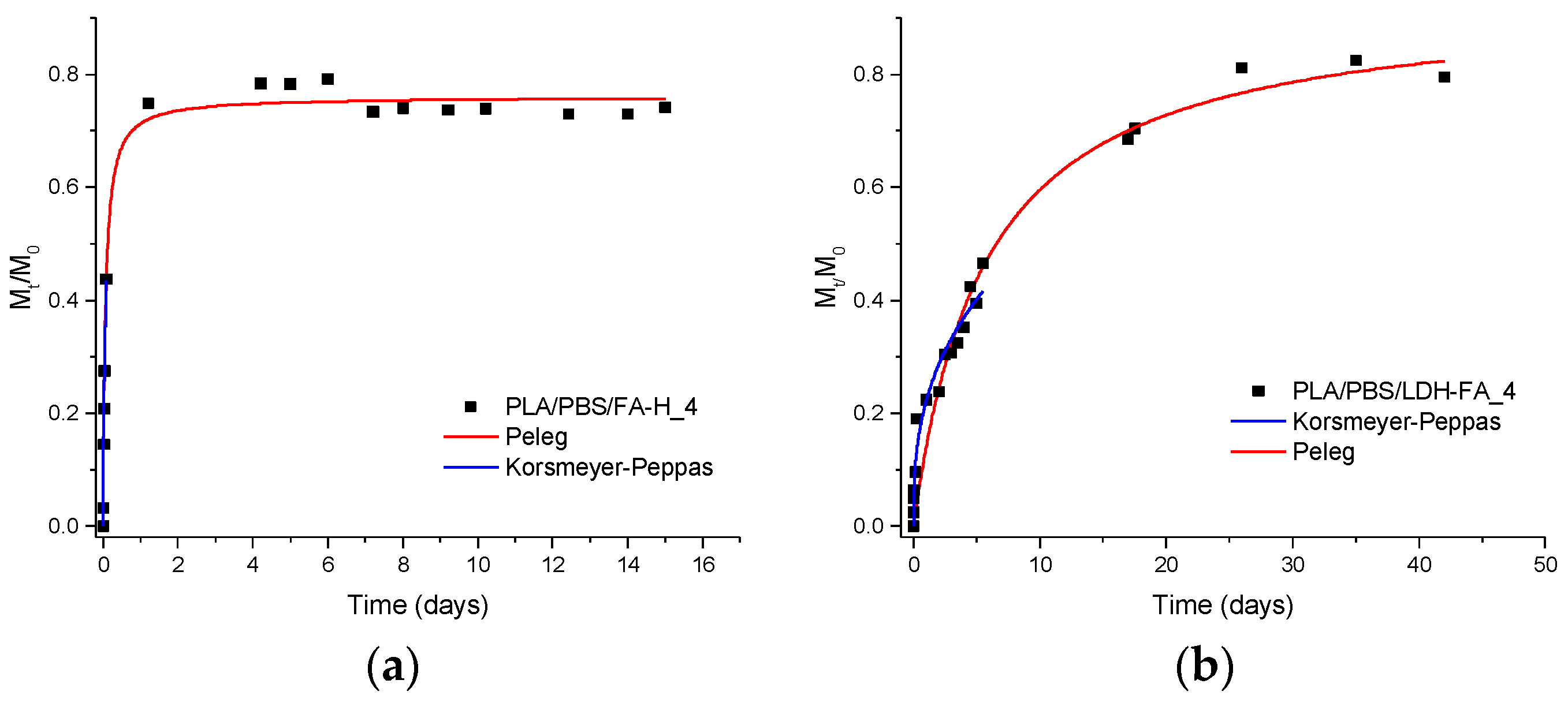
3.2.6. Migration Tests and Release Kinetics
3.3. PLA/PBSA/LDH Characterization via Micro-Compounder
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ncube, L.K.; Ude, A.U.; Ogunmuyiwa, E.N.; Zulkifli, R.; Beas, I.N. Environmental Impact of Food Packaging Materials: A Review of Contemporary Development from Conventional Plastics to Polylactic Acid Based Materials. Materials 2020, 13, 4994. [Google Scholar] [CrossRef] [PubMed]
- Tawakkal, I.S.M.A.; Cran, M.J.; Miltz, J.; Bigger, S.W. A Review of Poly(Lactic Acid)-Based Materials for Antimicrobial Packaging. J. Food Sci. 2014, 79, R1477–R1490. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.E. Beyond Plastic Waste. Science 2017, 358, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Westlake, J.R.; Tran, M.W.; Jiang, Y.; Zhang, X.; Burrows, A.D.; Xie, M. Biodegradable Active Packaging with Controlled Release: Principles, Progress, and Prospects. ACS Food Sci. Technol. 2022, 2, 1166–1183. [Google Scholar] [CrossRef]
- Rosenboom, J.G.; Langer, R.; Traverso, G. Bioplastics for a Circular Economy. Nat. Rev. Mater. 2022, 7, 117–137. [Google Scholar] [CrossRef]
- Nanda, S.; Patra, B.R.; Patel, R.; Bakos, J.; Dalai, A.K. Innovations in Applications and Prospects of Bioplastics and Biopolymers: A Review. Environ. Chem. Lett. 2022, 20, 379–395. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A. Recycling of Bioplastic Waste: A Review. Adv. Ind. Eng. Polym. Res. 2021, 4, 159–177. [Google Scholar] [CrossRef]
- La Mantia, F.P.; Botta, L.; Mistretta, M.C.; Di Fiore, A.; Titone, V. Recycling of a Biodegradable Polymer Blend. Polymers 2020, 12, 2297. [Google Scholar] [CrossRef]
- Swetha, T.A.; Bora, A.; Mohanrasu, K.; Balaji, P.; Raja, R.; Ponnuchamy, K.; Muthusamy, G.; Arun, A. A Comprehensive Review on Polylactic Acid (PLA)—Synthesis, Processing and Application in Food Packaging. Int. J. Biol. Macromol. 2023, 234, 123715. [Google Scholar] [CrossRef]
- Shahdan, D.; Rosli, N.A.; Chen, R.S.; Ahmad, S.; Gan, S. Strategies for Strengthening Toughened Poly(Lactic Acid) Blend via Natural Reinforcement with Enhanced Biodegradability: A Review. Int. J. Biol. Macromol. 2023, 251, 126214. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, J.; Li, J.; Liang, X.; Zhou, W.; Peng, S. Strategies and Techniques for Improving Heat Resistance and Mechanical Performances of Poly(Lactic Acid) (PLA) Biodegradable Materials. Int. J. Biol. Macromol. 2022, 218, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Lee, J. Comparison of Recently Developed Toughening Strategies for Polylactic Acid Blends. J. Ind. Eng. Chem. 2023, 125, 50–57. [Google Scholar] [CrossRef]
- Arrigo, R.; D’Anna, A.; Frache, A. Fully Bio-Based Ternary Polymer Blends: Structural Characterization and Mechanical Behavior. Mater. Today Sustain. 2023, 21, 100314. [Google Scholar] [CrossRef]
- Palai, B.; Mohanty, S.; Nayak, S.K. Synergistic Effect of Polylactic Acid(PLA) and Poly(Butylene Succinate-Co-Adipate) (PBSA) Based Sustainable, Reactive, Super Toughened Eco-Composite Blown Films for Flexible Packaging Applications. Polym. Test. 2020, 83, 106130. [Google Scholar] [CrossRef]
- Fortelny, I.; Ujcic, A.; Fambri, L.; Slouf, M. Phase Structure, Compatibility, and Toughness of PLA/PCL Blends: A Review. Front. Mater. 2019, 6, 206. [Google Scholar] [CrossRef]
- Aversa, C.; Barletta, M.; Cappiello, G.; Gisario, A. Compatibilization Strategies and Analysis of Morphological Features of Poly(Butylene Adipate-Co-Terephthalate) (PBAT)/Poly(Lactic Acid) PLA Blends: A State-of-Art Review. Eur. Polym. J. 2022, 173, 111304. [Google Scholar] [CrossRef]
- Su, S.; Kopitzky, R.; Tolga, S.; Kabasci, S. Polylactide (PLA) and Its Blends with Poly(Butylene Succinate) (PBS): A Brief Review. Polymers 2019, 11, 1193. [Google Scholar] [CrossRef]
- Zhao, T.; Yu, J.; Pan, H.; Zhao, Y.; Zhang, Q.; Yu, X.; Bian, J.; Han, L.; Zhang, H. Super-Tough Polylactic Acid (PLA)/Poly(Butylene Succinate) (PBS) Materials Prepared through Reactive Blending with Epoxy-Functionalized PMMA-GMA Copolymer. Int. J. Biol. Macromol. 2023, 251, 126150. [Google Scholar] [CrossRef]
- Ullah, M.S.; Yildirim, R.; Kodal, M.; Ozkoc, G. Reactive Compatibilization of PLA/PBS Bio-Blends via a New Generation of Hybrid Nanoparticles. J. Vinyl Addit. Technol. 2022, 29, 737–757. [Google Scholar] [CrossRef]
- Platnieks, O.; Gaidukovs, S.; Thakur, V.K.; Barkane, A.; Beluns, S. Bio-Based Poly (Butylene Succinate): Recent Progress, Challenges and Future Opportunities. Eur. Polym. J. 2021, 161, 110855. [Google Scholar] [CrossRef]
- Coltelli, M.B.; Aliotta, L.; Fasano, G.; Miketa, F.; Brkić, F.; Alonso, R.; Romei, M.; Cinelli, P.; Canesi, I.; Gigante, V.; et al. Recyclability Studies on Poly(lactic acid)/Poly(butylene succinate-co-adipate) (PLA/PBSA) Biobased and Biodegradable Films. Macromol. Mater. Eng. 2023, 2300136. [Google Scholar] [CrossRef]
- Aliotta, L.; Gigante, V.; Dal Pont, B.; Miketa, F.; Coltelli, M.-B.; Lazzeri, A. Tearing Fracture of Poly(Lactic Acid) (PLA)/ Poly(Butylene Succinate-Co-Adipate) (PBSA) Cast Extruded Films: Effect of the PBSA Content. Eng. Fract. Mech. 2023, 289, 109450. [Google Scholar] [CrossRef]
- Coiai, S.; Di Lorenzo, M.L.; Cinelli, P.; Righetti, M.C.; Passaglia, E. Binary Green Blends of Poly(lactic acid) with Poly(butylene adipate-co-butylene terephthalate) and Poly(butylene succinate-co-butylene adipate) and Their Nanocomposites. Polymers 2021, 13, 2489. [Google Scholar] [CrossRef] [PubMed]
- Changwichan, K.; Silalertruksa, T.; Gheewala, S.H. Eco-Efficiency Assessment of Bioplastics Production Systems and End-of-Life Options. Sustainability 2018, 10, 952. [Google Scholar] [CrossRef]
- Nettleton, D.F.; Fernandez-Avila, C.; Sanchez-Esteva, S.; Verstichel, S.; Coltelli, M.B.; Marti-Soler, H.; Aliotta, L.; Gigante, V. Biodegradation Prediction and Modelling for Decision Support. In Proceedings of the 12th International Conference on Simulation and Modeling Methodologies, Technologies and Applications, Lisbon, Portugal, 14–16 July 2022; pp. 26–35. [Google Scholar]
- Aliotta, L.; Vannozzi, A.; Canesi, I.; Cinelli, P.; Coltelli, M.B.; Lazzeri, A. Poly(Lactic Acid) (PLA)/Poly(Butylene Succinate-Co-Adipate) (PBSA) Compatibilized Binary Biobased Blends: Melt Fluidity, Morphological, Thermo-Mechanical and Micromechanical Analysis. Polymers 2021, 13, 218. [Google Scholar] [CrossRef]
- Messin, T.; Follain, N.; Guinault, A.; Sollogoub, C.; Gaucher, V.; Delpouve, N.; Marais, S. Structure and Barrier Properties of Multinanolayered Biodegradable PLA/PBSA Films: Confinement Effect via Forced Assembly Coextrusion. ACS Appl. Mater. Interfaces 2017, 9, 29101–29112. [Google Scholar] [CrossRef]
- Suwanamornlert, P.; Kerddonfag, N.; Sane, A.; Chinsirikul, W.; Zhou, W.; Chonhenchob, V. Poly(Lactic Acid)/Poly(Butylene-Succinate-Co-Adipate) (PLA/PBSA) Blend Films Containing Thymol as Alternative to Synthetic Preservatives for Active Packaging of Bread. Food Packag. Shelf Life 2020, 25, 100515–100517. [Google Scholar] [CrossRef]
- Panariello, L.; Coltelli, M.-B.; Vannozzi, A.; Bonacchi, D.; Aliotta, L.; Lazzeri, A. Fully Biobased Reactive Extrusion of Biocomposites Based on PLA Blends and Hazelnut Shell Powders (HSP). Chemistry 2021, 3, 1464–1480. [Google Scholar] [CrossRef]
- Stanley, J.; John, A.; Pušnik Črešnar, K.; Fras Zemljič, L.; Lambropoulou, D.A.; Bikiaris, D.N. Active Agents Incorporated in Polymeric Substrates to Enhance Antibacterial and Antioxidant Properties in Food Packaging Applications. Macromol 2022, 3, 1–27. [Google Scholar] [CrossRef]
- Ordoñez, R.; Atarés, L.; Chiralt, A. Biodegradable Active Materials Containing Phenolic Acids for Food Packaging Applications. Compr. Rev. Food Sci. Food Saf. 2022, 21, 3910–3930. [Google Scholar] [CrossRef]
- Zduńska, K.; Dana, A.; Kolodziejczak, A.; Rotsztejn, H. Antioxidant Properties of Ferulic Acid and Its Possible Application. Skin. Pharmacol. Physiol. 2018, 31, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Reano, A.F.; Domenek, S.; Pernes, M.; Beaugrand, J.; Allais, F. Ferulic Acid-Based Bis/Trisphenols as Renewable Antioxidants for Polypropylene and Poly(Butylene Succinate). ACS Sustain. Chem. Eng. 2016, 4, 6562–6571. [Google Scholar] [CrossRef]
- Ordoñez, R.; Atarés, L.; Chiralt, A. Antilisterial Action of PLA Films with Ferulic Acid as Affected by the Method of Incorporation. Food Biosci. 2022, 49, 101865. [Google Scholar] [CrossRef]
- Pernin, A.; Bosc, V.; Maillard, M.N.; Dubois-Brissonnet, F. Ferulic Acid and Eugenol Have Different Abilities to Maintain Their Inhibitory Activity against Listeria Monocytogenes in Emulsified Systems. Front. Microbiol. 2019, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Aragón-Gutiérrez, A.; Rosa, E.; Gallur, M.; López, D.; Hernández-Muñoz, P.; Gavara, R. Melt-Processed Bioactive Evoh Films Incorporated with Ferulic Acid. Polymers 2021, 13, 68. [Google Scholar] [CrossRef]
- Ordoñez, R.; Atarés, L.; Chiralt, A. Antibacterial Properties of Cinnamic and Ferulic Acids Incorporated to Starch and PLA Monolayer and Multilayer Films. Food Control 2022, 136, 108878. [Google Scholar] [CrossRef]
- Sharma, S.; Jaiswal, A.K.; Duffy, B.; Jaiswal, S. Ferulic Acid Incorporated Active Films Based on Poly(Lactide)/Poly(Butylene Adipate-Co-Terephthalate) Blend for Food Packaging. Food Packag. Shelf Life 2020, 24, 100491. [Google Scholar] [CrossRef]
- Kahya, N.; Kestir, S.M.; Öztürk, S.; Yolaç, A.; Torlak, E.; Kalaycıoğlu, Z.; Akın-Evingür, G.; Erim, F.B. Antioxidant and Antimicrobial Chitosan Films Enriched with Aqueous Sage and Rosemary Extracts as Food Coating Materials: Characterization of the Films and Detection of Rosmarinic Acid Release. Int. J. Biol. Macromol. 2022, 217, 470–480. [Google Scholar] [CrossRef]
- Kowalska, A.; Kalinowska-Lis, U. 18β-Glycyrrhetinic Acid: Its Core Biological Properties and Dermatological Applications. Int. J. Cosmet. Sci. 2019, 41, 325–331. [Google Scholar] [CrossRef]
- Darvishi, B.; Manoochehri, S.; Kamalinia, G.; Samadi, N.; Amini, M.; Mostafavi, S.H.; Maghazei, S.; Atyabi, F.; Dinarvand, R. Preparation and Antibacterial Activity Evaluation of 18-β-Glycyrrhetinic Acid Loaded PLGA Nanoparticles. Iran. J. Pharm. Res. 2015, 14, 373–383. [Google Scholar] [CrossRef]
- Miletić, A.; Ristić, I.; Coltelli, M.B.; Pilić, B. Modification of PLA-Based Films by Grafting or Coating. J. Funct. Biomater. 2020, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, A.; Delshadi, R.; Assadpour, E.; Jafari, S.M.; Williams, L. Antimicrobial-Loaded Nanocarriers for Food Packaging Applications. Adv. Colloid Interface Sci. 2020, 278, 102140. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Sit, N. Application of Natural Extracts as Active Ingredient in Biopolymer Based Packaging Systems. J. Food Sci. Technol. 2023, 60, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Costantino, U.; Ambrogi, V.; Nocchetti, M.; Perioli, L. Hydrotalcite-like Compounds: Versatile Layered Hosts of Molecular Anions with Biological Activity. Microporous Mesoporous Mater. 2008, 107, 149–160. [Google Scholar] [CrossRef]
- Mishra, G.; Dash, B.; Pandey, S. Layered Double Hydroxides: A Brief Review from Fundamentals to Application as Evolving Biomaterials. Appl. Clay Sci. 2018, 153, 172–186. [Google Scholar] [CrossRef]
- Cicogna, F.; Passaglia, E.; Benedettini, M.; Oberhauser, W.; Ishak, R.; Signori, F.; Coiai, S. Rosmarinic and Glycyrrhetinic Acid-Modified Layered Double Hydroxides as Functional Additives for Poly(Lactic Acid)/Poly(Butylene Succinate) Blends. Molecules 2023, 28, 347. [Google Scholar] [CrossRef]
- Zhong, G.; Qiu, M.; Zhang, J.; Jiang, F.; Yue, X.; Huang, C.; Zhao, S.; Zeng, R.; Zhang, C.; Qu, Y. Fabrication and Characterization of PVA@PLA Electrospinning Nanofibers Embedded with Bletilla Striata Polysaccharide and Rosmarinic Acid to Promote Wound Healing. Int. J. Biol. Macromol. 2023, 234, 123693. [Google Scholar] [CrossRef]
- Tammaro, L.; Vittoria, V.; Bugatti, V. Dispersion of Modified Layered Double Hydroxides in Poly(Ethylene Terephthalate) by High Energy Ball Milling for Food Packaging Applications. Eur. Polym. J. 2014, 52, 172–180. [Google Scholar] [CrossRef]
- Gorrasi, G.; Bugatti, V.; Vertuccio, L.; Vittoria, V.; Pace, B.; Cefola, M.; Quintieri, L.; Bernardo, P.; Clarizia, G. Active Packaging for Table Grapes: Evaluation of Antimicrobial Performances of Packaging for Shelf Life of the Grapes under Thermal Stress. Food Packag. Shelf Life 2020, 25, 100545. [Google Scholar] [CrossRef]
- Coiai, S.; Cicogna, F.; Pinna, S.; Spiniello, R.; Onor, M.; Oberhauser, W.; Coltelli, M.B.; Passaglia, E. Antibacterial LDPE-Based Nanocomposites with Salicylic and Rosmarinic Acid-Modified Layered Double Hydroxides. Appl. Clay Sci. 2021, 214, 106276. [Google Scholar] [CrossRef]
- Rossi, C.; Schoubben, A.; Ricci, M.; Perioli, L.; Ambrogi, V.; Latterini, L.; Aloisi, G.G.; Rossi, A. Intercalation of the Radical Scavenger Ferulic Acid in Hydrotalcite-like Anionic Clays. Int. J. Pharm. 2005, 295, 47–55. [Google Scholar] [CrossRef]
- Wu, J.; Lu, J.; Hu, J.; Gao, Y.; Ma, Q.; Ju, Y. Self-Assembly of Sodium Glycyrrhetinate into a Hydrogel: Characterisation and Properties. RSC Adv. 2013, 3, 24906–24909. [Google Scholar] [CrossRef]
- Bastianini, M.; Faffa, C.; Sisani, M.; Petracci, A. Caffeic Acid-Layered Double Hydroxide Hybrid: A New Raw Material for Cosmetic Applications. Cosmetics 2018, 5, 51. [Google Scholar] [CrossRef]
- Peleg, M. An Empirical Model for the Description of Moisture Sorption Curves. J. Food Sci. 1988, 53, 1216–1217. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Higuchi Equation: Derivation, Applications, Use and Misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, H.J.; Yang, J.H.; Kim, T.H.; Choi, G.; Paek, S.M.; Choi, A.J.; Choy, J.H.; Oh, J.M. Intracrystalline Structure and Release Pattern of Ferulic Acid Intercalated into Layered Double Hydroxide through Various Synthesis Routes. Appl. Clay Sci. 2015, 112–113, 32–39. [Google Scholar] [CrossRef]
- Muksing, N.; Magaraphan, R.; Coiai, S.; Passaglia, E. Effect of Surfactant Alkyl Chain Length on the Dispersion, and Thermal and Dynamic Mechanical Properties of LDPE/Organo-LDH Composites. Express Polym. Lett. 2011, 5, 428–448. [Google Scholar] [CrossRef]
- Kalinowska, M.; Piekut, J.; Bruss, A.; Follet, C.; Sienkiewicz-Gromiuk, J.; Świsłocka, R.; Rzaczyńska, Z.; Lewandowski, W. Spectroscopic (FT-IR, FT-Raman, 1H, 13C NMR, UV/VIS), Thermogravimetric and Antimicrobial Studies of Ca(II), Mn(II), Cu(II), Zn(II) and Cd(II) Complexes of Ferulic Acid. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 122, 631–638. [Google Scholar] [CrossRef]
- Righetti, M.C.; Gazzano, M.; Di Lorenzo, M.L.; Androsch, R. Enthalpy of Melting of α′- and α-Crystals of Poly(L-Lactic Acid). Eur. Polym. J. 2015, 70, 215–220. [Google Scholar] [CrossRef]
- Pan, P.; Zhu, B.; Kai, W.; Dong, T.; Inoue, Y. Polymorphic Transition in Disordered Poly(L-Lactide) Crystals Induced by Annealing at Elevated Temperatures. Macromolecules 2008, 41, 4296–4304. [Google Scholar] [CrossRef]
- Yoo, E.S.; Im, S.S. Melting Behavior of Poly(Butylene Succinate) during Heating Scan by DSC. J. Polym. Sci. B Polym. Phys. 1999, 37, 1357–1366. [Google Scholar] [CrossRef]
- Zou, H.; Yi, C.; Wang, L.; Liu, H.; Xu, W. Thermal Degradation of Poly(Lactic Acid) Measured by Thermogravimetry Coupled to Fourier Transform Infrared Spectroscopy. J. Therm. Anal. Calorim. 2009, 97, 929–935. [Google Scholar] [CrossRef]
- Strangis, G.; Rossi, D.; Cinelli, P.; Seggiani, M. Seawater Biodegradable Poly(Butylene Succinate-Co-Adipate)—Wheat Bran Biocomposites. Materials 2023, 16, 2593. [Google Scholar] [CrossRef]
- Oliveira, M.; Santos, E.; Araújo, A.; Fechine, G.J.M.; Machado, A.V.; Botelho, G. The Role of Shear and Stabilizer on PLA Degradation. Polym. Test. 2016, 51, 109–116. [Google Scholar] [CrossRef]
- Coiai, S.; Cicogna, F.; de Santi, A.; Pérez Amaro, L.; Spiniello, R.; Signori, F.; Fiori, S.; Oberhauser, W.; Passaglia, E. MMT and LDH Organo-Modification with Surfactants Tailored for PLA Nanocomposites. Express Polym. Lett. 2017, 11, 163–175. [Google Scholar] [CrossRef]
- Georgousopoulou, I.N.; Vouyiouka, S.; Dole, P.; Papaspyrides, C.D. Thermo-Mechanical Degradation and Stabilization of Poly(Butylene Succinate). Polym. Degrad. Stab. 2016, 128, 182–192. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, Y.; Duan, J.; Yang, J.; Wang, Y.; Zhang, C. Accelerated Hydrolytic Degradation of Poly(Lactic Acid) Achieved by Adding Poly(Butylene Succinate). Polym. Bull. 2016, 73, 1067–1083. [Google Scholar] [CrossRef]
- Somsunan, R.; Noppakoon, S.; Punyodom, W. Effect of G40 Plasticizer on the Properties of Ternary Blends of Biodegradable PLA/PBS/G40. J. Polym. Res. 2019, 26, 92. [Google Scholar] [CrossRef]
- Bhatia, A.; Gupta, R.K.; Bhattacharya, S.N.; Choi, H.J. Analysis of Gas Permeability Characteristics of Poly(Lactic Acid)/Poly(Butylene Succinate) Nanocomposites. J. Nanomater. 2012, 2012, 249094. [Google Scholar] [CrossRef]
- EUR-Lex—32011R0010. Available online: http://data.europa.eu/eli/reg/2011/10/oj (accessed on 28 September 2023).
- Ordoñez, R.; Atarés, L.; Chiralt, A. Effect of Ferulic and Cinnamic Acids on the Functional and Antimicrobial Properties in Thermo-Processed PLA Films. Food Packag. Shelf Life 2022, 33, 100882. [Google Scholar] [CrossRef]
- Miyagusuku-Cruzado, G.; García-Cano, I.; Rocha-Mendoza, D.; Jiménez-Flores, R.; Giusti, M.M. Monitoring Hydroxycinnamic Acid Decarboxylation by Lactic Acid Bacteria Using High-Throughput UV-Vis Spectroscopy. Molecules 2020, 25, 3142. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Lee, J.Y.; Ahn, S.J.; Choi, H.J. Melt Rheology and Mechanical Characteristics of Poly(Lactic Acid)/Alkylated Graphene Oxide Nanocomposites. Polymers 2020, 12, 2402. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, D.; Wan, G.; Li, B.; Zhao, G. Glass Fiber Reinforced PLA Composite with Enhanced Mechanical Properties, Thermal Behavior, and Foaming Ability. Polymer 2019, 181, 121803. [Google Scholar] [CrossRef]
- Tian, J.; Cao, Z.; Qian, S.; Xia, Y.; Zhang, J.; Kong, Y.; Sheng, K.; Zhang, Y.; Wan, Y.; Takahashi, J. Improving Tensile Strength and Impact Toughness of Plasticized Poly(Lactic Acid) Biocomposites by Incorporating Nanofibrillated Cellulose. Nanotechnol. Rev. 2022, 11, 2469–2482. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tonset a (°C) | Tmax (PLA) b (°C) | Tmax (PBSA) b (°C) |
|---|---|---|---|
| PLA/PBSA | 332 | 365 | 397 |
| PLA/PBSA/LDH-FA_1 | 300 | 336 | 394 |
| PLA/PBSA/LDH-FA_4 | 289 | 322 | 395 |
| PLA/PBSA/LDH-RA_1 | 331 | 358 | 392 |
| PLA/PBSA/LDH-RA_4 | 325 | 351 | 393 |
| PLA/PBSA/LDH-GA_1 | 295 | 329 | 394 |
| PLA/PBSA/LDH-GA_4 | 277 | 308 | 394 |
| Sample | Mn (g/mol) | Mw (g/mol) | Ð a |
|---|---|---|---|
| PLA from PLA/PBSA | 101,400 | 164,400 | 1.6 |
| PLA from PLA/PBSA/LDH-FA_4 | 65,000 | 116,000 | 1.8 |
| PLA from PLA/PBSA/LDH-RA_4 | 104,000 | 157,500 | 1.5 |
| PLA from PLA/PBSA/LDH-GA_4 | 75,000 | 120,000 | 1.6 |
| PBSA from PLA/PBSA | 53,600 | 114,000 | 2.1 |
| PBSA from PLA/PBSA/LDH-FA_4 | 48,000 | 95,000 | 2.0 |
| PBSA from PLA/PBSA/LDH-RA_4 | 55,000 | 99,000 | 1.8 |
| PBSA from PLA/PBSA/LDH-GA_4 | 53,000 | 90,000 | 1.7 |
| Sample | EC50 (µM) a |
|---|---|
| FA-H | 19 ± 1 |
| LDH-FA b | 17 ± 1 |
| RA-H | 2.7 ± 0.1 |
| LDH-RA b | 19 ± 7 |
| Trolox | 23 ± 3 |
| Sample | WVTR (g/m2 × 24 h) a |
|---|---|
| PLA | 13.5 |
| PLA/PBSA | 19.0 |
| PLA/PBSA/LDH-FA_1 | 18.5 |
| PLA/PBSA/LDH-FA_4 | 17.8 |
| PLA/PBSA/LDH-RA_1 | 13.4 |
| PLA/PBSA/LDH-RA_4 | 24.1 |
| PLA/PBSA/LDH-GA_1 | 17.0 |
| PLA/PBSA/LDH-GA_4 | 16.7 |
| Sample | Peleg | Korsmeyer-Peppas | |||||
|---|---|---|---|---|---|---|---|
| 1/k1 a (mg/Day) | M∞ b (mg/100 mg Film) | M∞/M0 c (%) | R2 d | n e | k f (Day−n) | R2 d | |
| PLA/PBSA/RA-H_4 | 20 ± 2 | 1.43 ± 0.02 | 77 ± 1 | 0.99 | 0.47 ± 0.06 | 1.6 ± 0.2 | 0.97 |
| PLA/PBSA/LDH-RA_4 | 0.023 ± 0.002 | 0.242 ± 0.007 | 13.2 ± 0.4 | 0.99 | 0.44 ± 0.01 | 0.19 ± 0.01 | 0.99 |
| PLA/PBSA/GA-H_4 | 7 ± 1 | 0.367 ± 0.008 | 64 ± 1 | 0.96 | 0.26 ± 0.01 | 1.05 ± 0.02 | 0.99 |
| PLA/PBSA/LDH-GA_4 | 0.28 ± 0.07 | 0.40 ± 0.02 | 70.1 ± 0.5 | 0.89 | 0.28 ± 0.04 | 0.48 ± 0.01 | 0.91 |
| Sample | Young’s Modulus (GPa) | Yield Stress (MPa) | Stress at Break (%) | Elongation at Break (%) |
|---|---|---|---|---|
| PLA/PBSA_100 | 1.8 ± 0.1 | 38.1 ± 0.2 | 29.9 ± 0.5 | 240 ± 41 |
| PLA/PBSA/LDH-RA_1_100 | 1.9 ± 0.2 | 34 ± 1 | 27 ± 1 | 206 ± 67 |
| PLA/PBSA/150 | 2.0 ± 0.2 | 40 ± 1 | 29.9 ± 0.2 | 244 ± 41 |
| PLA/PBSA/LDH-RA_1_150 | 2.1 ± 0.2 | 38 ± 1 | 27.0 ± 0.3 | 227 ± 48 |
| Sample | Mn (g/mol) | Mw (g/mol) | Ð 1 |
|---|---|---|---|
| PLA from PLA/PBSA_100 | 76,500 | 149,000 | 1.9 |
| PLA from PLA/PBSA_150 | 78,000 | 155,000 | 2.0 |
| PLA from PLA/PBSA/LDH-RA_1_100 | 66,300 | 145,500 | 2.2 |
| PLA from PLA/PBSA/LDH-RA_1_150 | 62,700 | 138,000 | 2.2 |
| PBSA from PLA/PBSA_100 | 59,000 | 127,000 | 2.2 |
| PBSA from PLA/PBSA_150 | 53,700 | 128,000 | 2.4 |
| PBSA from PLA/PBSA/LDH-RA_1_100 | 57,000 | 117,000 | 2.1 |
| PBSA from PLA/PBSA/LDH-RA_1_150 | 57,000 | 118,000 | 2.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cicogna, F.; Passaglia, E.; Telleschi, A.; Oberhauser, W.; Coltelli, M.-B.; Panariello, L.; Gigante, V.; Coiai, S. New Functional Bionanocomposites by Combining Hybrid Host-Guest Systems with a Fully Biobased Poly(lactic acid)/Poly(butylene succinate-co-adipate) (PLA/PBSA) Binary Blend. J. Funct. Biomater. 2023, 14, 549. https://0-doi-org.brum.beds.ac.uk/10.3390/jfb14110549
Cicogna F, Passaglia E, Telleschi A, Oberhauser W, Coltelli M-B, Panariello L, Gigante V, Coiai S. New Functional Bionanocomposites by Combining Hybrid Host-Guest Systems with a Fully Biobased Poly(lactic acid)/Poly(butylene succinate-co-adipate) (PLA/PBSA) Binary Blend. Journal of Functional Biomaterials. 2023; 14(11):549. https://0-doi-org.brum.beds.ac.uk/10.3390/jfb14110549
Chicago/Turabian StyleCicogna, Francesca, Elisa Passaglia, Alice Telleschi, Werner Oberhauser, Maria-Beatrice Coltelli, Luca Panariello, Vito Gigante, and Serena Coiai. 2023. "New Functional Bionanocomposites by Combining Hybrid Host-Guest Systems with a Fully Biobased Poly(lactic acid)/Poly(butylene succinate-co-adipate) (PLA/PBSA) Binary Blend" Journal of Functional Biomaterials 14, no. 11: 549. https://0-doi-org.brum.beds.ac.uk/10.3390/jfb14110549