1. Introduction
The last few decades have seen a revolutionary era in the field of drug delivery, leading to the development of several innovative cutting-edge approaches for diagnosis and treatment of many pathological states. Among the wide plethora of available approaches, the use of carriers as drug delivery vehicles has received a huge interest, overcoming many drawbacks of the traditional systemic administration route. In summary, drug transportation by specific carriers improves pharmacokinetic profiles, reduces the risk of side effects, and allows a localized and sustained payload release in the target area, with consequent advantages in terms of the required administered dosage to achieve the desired therapeutic effect. In addition, such carriers can be properly engineered to pass through many biological barriers and actively target a specific pathological area of the body. Micro- and nano-particles are among the most widely investigated drug carriers in nanomedicine. They are available in different sizes, forms and materials, such as synthetic polymers, proteins, lipids and inorganic materials. Among them, mesoporous particles hold huge potential in nanomedicine, due to their high versatility, allowing a fine-tuning of their physico-chemical and structural properties, the high loading capacity for many different payloads (e.g., hydrophilic and hydrophobic drugs, growth factors, microRNAs) and their potential for controlled release of their cargo. In particular, the use of mesoporous silica nanoparticles (MSNs) has experienced a great upsurge over the last decade, as recently thoroughly reviewed by Narayan and co-workers [
1] and Manzano and Vallet-Regì [
2]. For instance, Saini et al. recently reported on the design of MSNs with tunable pore diameter to enhance the delivery of gemcitabine in pancreatic cancer treatment [
3], meanwhile Kong et al. developed curcumin-loaded MSNs as a potential cancer therapy with improved stability, anti-oxidant and antitumor activity as compared to the freely administered drug [
4]. Moreover, a huge effort is being devoted to the surface functionalization of MSNs with the aim to improve their dispersibility in aqueous media [
5], to favor their cellular internalization [
6], to provide them with additional features, such as anti-oxidant properties [
7] or mucoadhesiveness [
8], and to make them able to respond to external stimuli, thus triggering the release of their payload [
9,
10,
11,
12,
13,
14]. A further possibility to functionalize MSNs and afford additional bioactivity consists in doping their framework with other elements, which will be then released in the form of therapeutic ions, such as copper, strontium and calcium species, in a target area of the body [
15,
16,
17,
18,
19].
Among mesoporous materials, another class of particles, namely mesoporous carbon nanoparticles (MCNs), has also revealed interesting properties for drug delivery. Nevertheless, MCNs have been poorly investigated so far, despite their enormous potential and advantages over their silica-based counterparts. Indeed, MCNs combine the characteristic advantages of mesoporous particles with the benefits of carbonaceous materials: (i) favorable structural properties (e.g., high specific surface area and pore volume) for drug loading; (ii) high versatility to allow control over payload release kinetics; (iii) easy surface functionalization for an active targeting or a triggered payload release; (iv) excellent heat conversion capability for potential application in photothermal therapy; (v) high biocompatibility and chemical stability; and (vi) superior loading capacity of aromatic and poorly water soluble compounds [
20]. Representing a next generation of mesoporous materials for drug delivery, MCNs are currently experiencing a similar growth, as did MSNs in the past. Whereas MSNs require chemical agents (e.g., silanes, glutaraldehyde) or physical methods (e.g., laser treatment) for their surface functionalization [
21,
22], abundant functional groups (manly carboxyl groups) can be easily exposed on MCN surface through oxidation treatment [
20]. Similar to their silica counterparts, MCNs have also been precisely processed to provide them with specific features. For instance, MCNs have been recently surface functionalized to achieve a pH-triggered payload release [
23], decorated with silver nanoparticles to design an anti-bacterial platform for wound healing applications [
24], and surface-modified to overcome mucous and epithelial biological barriers, thus improving drug bioavailability [
25]. Among the wide variety of mesoporous materials available for drug loading (e.g., MCNs, MSNs, mesoporous hydroxyapatite and mesoporous metallic oxide), the low density, high porosity and strong adsorption ability of MCNs make them able to host huge amounts of drugs (in particular hydrophobic drugs that usually suffer for poor bioavailability), which is a strict requisite for all those applications requiring high drug dosages [
20]. In addition, the high MCN drug loading capacity and their capability to generate a photothermal effect are currently under investigation to develop combined chemo-photothermal therapies for a non-invasive treatment of cancer [
26].
However, irrespective of their nature, a common issue that characterizes all particulate drug delivery systems is the need to selectively accumulate and retain them in the target tissue for the required time to allow a complete payload release at an effective concentration to exert its therapeutic function. In this regard, the encapsulation of drug-loaded particles into a hydrogel vehicle phase could represent an effective strategy to locally inject a predefined volume of a therapeutic formulation, which will be then in situ retained for a predefined time interval, while progressively releasing the payload. For instance, polymeric particles have been loaded into hydrogels to achieve a sustained and prolonged release of neuroprotective and neuroregenerative drugs [
27], growth factors (i.e., stromal cell-derived factor-1, vascular endothelial growth factor) [
28], insulin [
29,
30] and platinum compounds for cancer treatment [
31]. MSN-hydrogel hybrid formulations have been also widely explored for the delivery of drugs/biomacromolecules for cartilage regeneration [
32], curcumin for dermal application [
33], ibuprofen as an anti-inflammatory drug [
34,
35] and therapeutic ions [
35,
36]. In addition, we have recently demonstrated that the encapsulation of pH-sensitive MSNs into thermosensitive hydrogels did not alter their responsiveness to external environmental changes (i.e., pH changes in the surrounding environment) [
37]. On the contrary, to the best of our knowledge there are no reports in the literature on MCN encapsulation into a hydrogel vehicle phase. Hence, the first aim of the present work was to investigate the loading of two ordered mesoporous carbons (OMCs), which have different geometrical and pore properties into polymeric thermosensitive sol-gel systems based on a customized poly(ether urethane) containing Poloxamer
® 407 as a building block [
24,
35,
36,
37]. Complete characterization of OMCs was performed through Scanning Electron Microscopy, Small Angle X-ray Scattering, N
2 adsorption/desorption measurements at 77K, Differential Scanning Calorimetry and Thermogravimetric Analysis, while the successful synthesis of the poly(ether urethane) was verified through Infrared spectroscopy, Nuclear Magnetic Resonance spectroscopy and Size Exclusion Chromatography. Hydrogels were prepared at 15 and 20%
w/v concentrations and loaded with OMCs at 10 mg/mL concentration. The effects of OMC loading on the gelling properties of the hybrid formulations were studied through qualitative tube inverting tests and rheological analyses. As a second step, the anti-inflammatory drug ibuprofen (IBU) was loaded into the OMCs through a melt infiltration method, leading to IBU-OMCs composites, which were encapsulated into the hydrogels and the resulting formulations were rheologically characterized. Lastly, the release profile of the drug in physiological-like conditions was assessed and compared to that measured from gel systems loaded with pristine ibuprofen molecules at the same concentration.
2. Materials and Methods
2.1. Synthesis of Ibuprofen-Loaded Ordered Mesoporous Carbons
2.1.1. Materials
The triblock copolymers EO20PO70EO20 (ethylene oxide-propylene oxide-ethylene oxide, Pluronic P123) and EO106PO70EO106 (Pluronic F127), cationic cetyltrimethylammonium bromide (CTAB) and tetraethyl orthosilicate (TEOS 98%) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used to synthesize mesoporous silicas, which were then employed as templates for OMC synthesis. Sucrose (≥99.5%, Sigma-Aldrich, St. Louis, MO, USA) was used as a carbon precursor during OMC synthesis. Ibuprofen (>98%) was purchased from Sigma-Aldrich (St. Louis, MO, USA). All solvents were also purchased from Sigma-Aldrich (St. Louis, MO, USA) in the analytical grade and used as received.
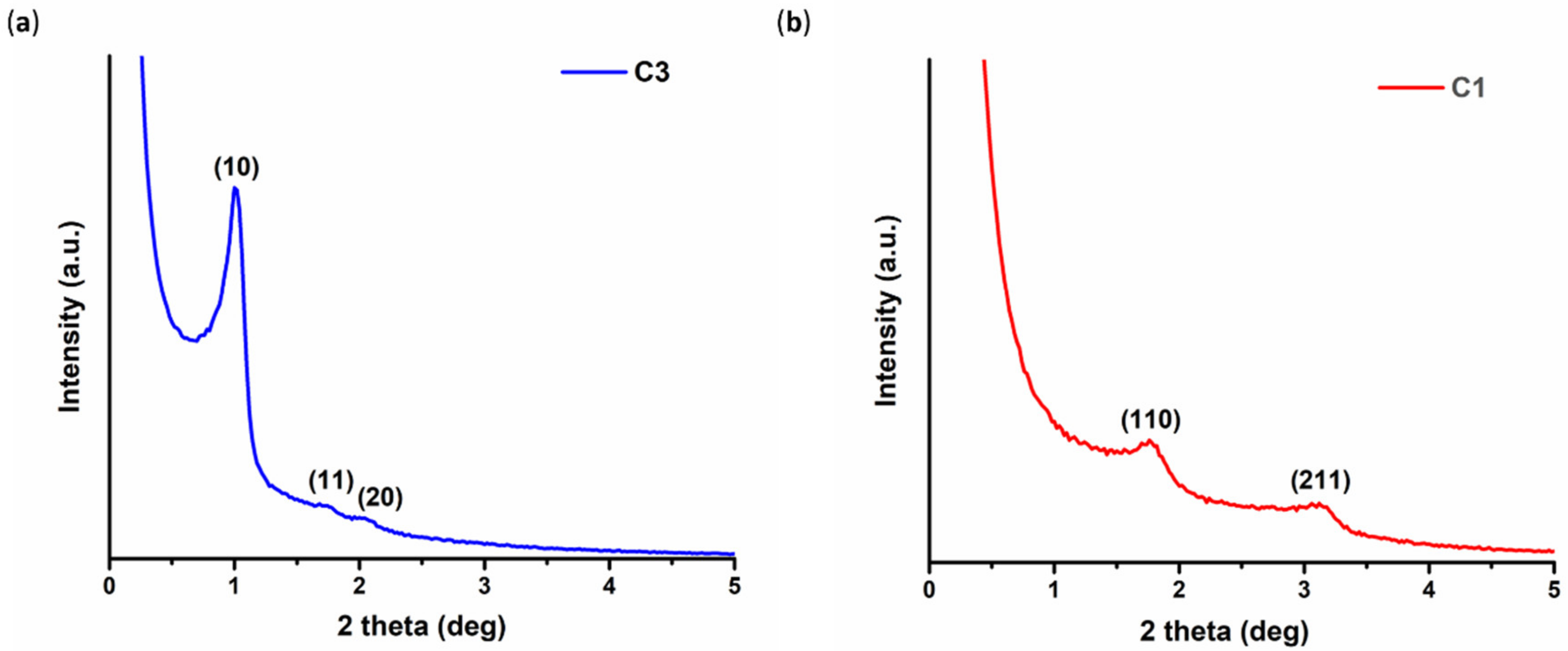
2.1.2. Rod-Like CMK-3 Type Ordered Mesoporous Carbons
The CMK-3–type carbon material (denoted hereafter as C3) was synthesized, starting from the hexagonally ordered mesoporous silica SBA-15 using a standard hard-templating approach [
38]. The SBA-15 silica template was prepared according to a typical method [
39], using Pluronic P123 as surfactant agent, and TEOS as a silica source at a composition of 4 g of P123:0.041 mol TEOS:6.67 mol H
2O:0.24 mol HCl. The hydrothermal reaction took place in an autoclave at 35 °C for 20 h and the aging process was conducted at 90 °C for 24 h. Aiming to obtain a carbon sample with the inverse structure, SBA-15 was impregnated twice with an acidic sucrose solution, subsequently polymerized at 100 °C and 160 °C in air for 6 h and pyrolyzed at 900 °C for at least 2 h in a tube furnace under N
2 gas flow (100 mL/min). The synthesis was concluded after etching of the silica framework using HF at room temperature (RT) and thorough washing of the remaining carbon with water and ethanol [
40].
2.1.3. Spherical CMK-1 Ordered Mesoporous Carbon
In a similar approach, CMK-1 carbon spheres (denoted hereafter as C1) were prepared using sucrose as a carbon precursor and MCM-48 spheres as a starting template. MCM-48 is a 3D cubic periodic silica with an interpenetrating network of pores, in contrast to the uni-dimensional hexagonal pores of SBA-15. Τhe MCM-48 silica spheres were prepared based on a modified Stöber method [
41], using a mixture of two surfactants (CTAB as porogen and Pluronic F127 as shape modulator), ethanol, ammonia, H
2O and TEOS at room temperature [
42]. The impregnation, polymerization and pyrolysis conditions were identical to the C3 case. The carbon with the inverse structure and the spherical morphology was obtained again by dissolution of the silica with hydrofluoric acid.
2.1.4. Drug-Loaded Ordered Mesoporous C3 and C1 Carbons
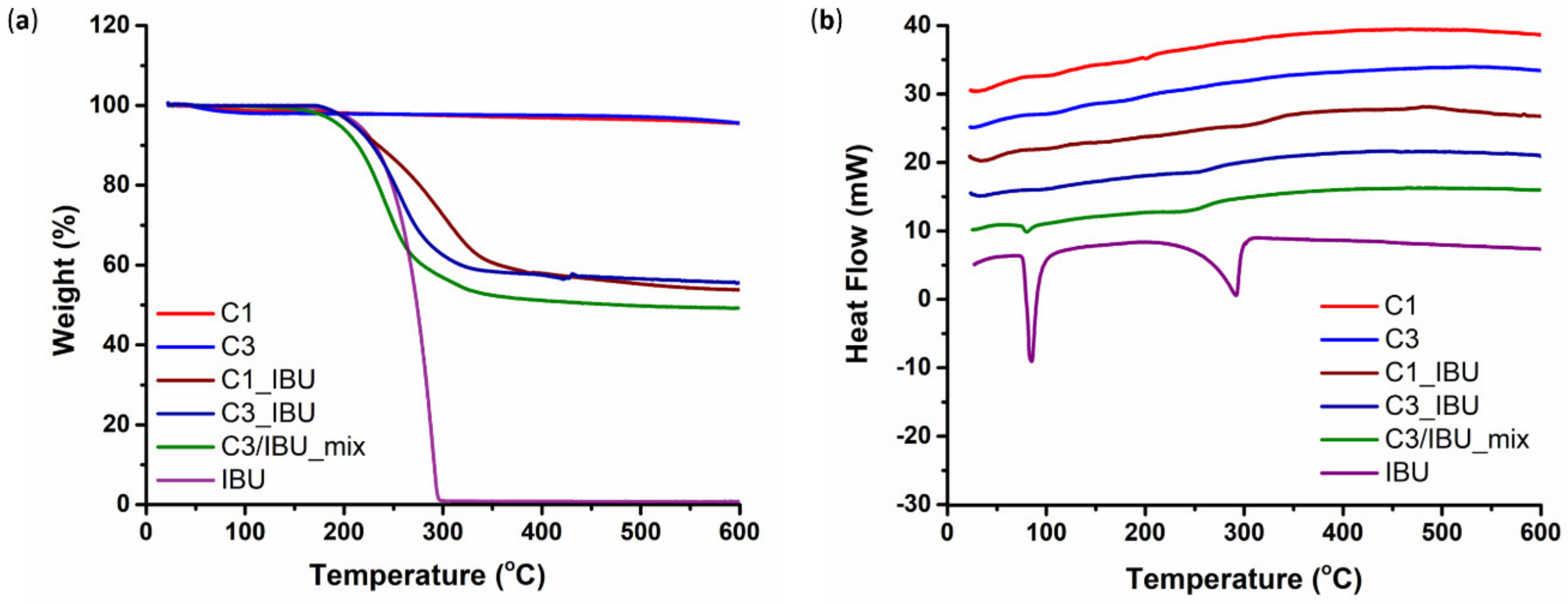
A melt infiltration method was used to incorporate ibuprofen (denoted hereafter as IBU) into the pores of the carbon particles (both C3 and C1). More specifically, C3 or C1 carbon and IBU powders were ground together to obtain a physical mixture with a 1:1 weight ratio, aiming to completely fill the total pore volume of the particles. The mixture was immersed in a water bath at 86–87 °C, i.e., above the melting point of IBU (75–78 °C), under rotation for 30 min, using a rotary evaporator (RII, BÜCHI AG, Flawil, Switzerland), so that the drug would melt and infiltrate the particles’ pores. Subsequently, the mixture was transferred to an ice bath and was kept there for 10 min to induce the solidification of IBU.
2.2. Characterization of C3 and C1 Based Materials
The surface morphology of the synthesized C3 and C1 carbon materials was studied through Scanning Electron Microscopy (SEM) using a JSM 7401F Field Emission Microscope (JEOL Ltd, Tokyo, Japan) equipped with a Gentle Beam mode.
The ordered pore structure of the samples was investigated using Small Angle X-ray Scattering (SAXS) in transmission mode on a SmartLab X-ray diffraction system (Rigaku Corporation, Tokyo, Japan) equipped with SAXS optics (λ = 1.54 Å). The scans were obtained from 0.06 to 8 degrees, with a speed of 0.1 deg/min and a step of 0.02 degrees.
The incorporation of IBU into the carbons’ pores was checked by Thermo-Gravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC). The measurements were performed on approximately 10 mg of each drug-loaded carbon sample, in the range of 25–600 °C, with a heating rate of 10 °C/min in an Al2O3 crucible under Argon flow (30 mL/min), using a SETSYS Evolution 18 Analyser (Setaram Instrumentation, Caluire-et-Cuire, France). Purging prior to thermal analysis as well as buoyancy corrections through blank measurements were also carried out.
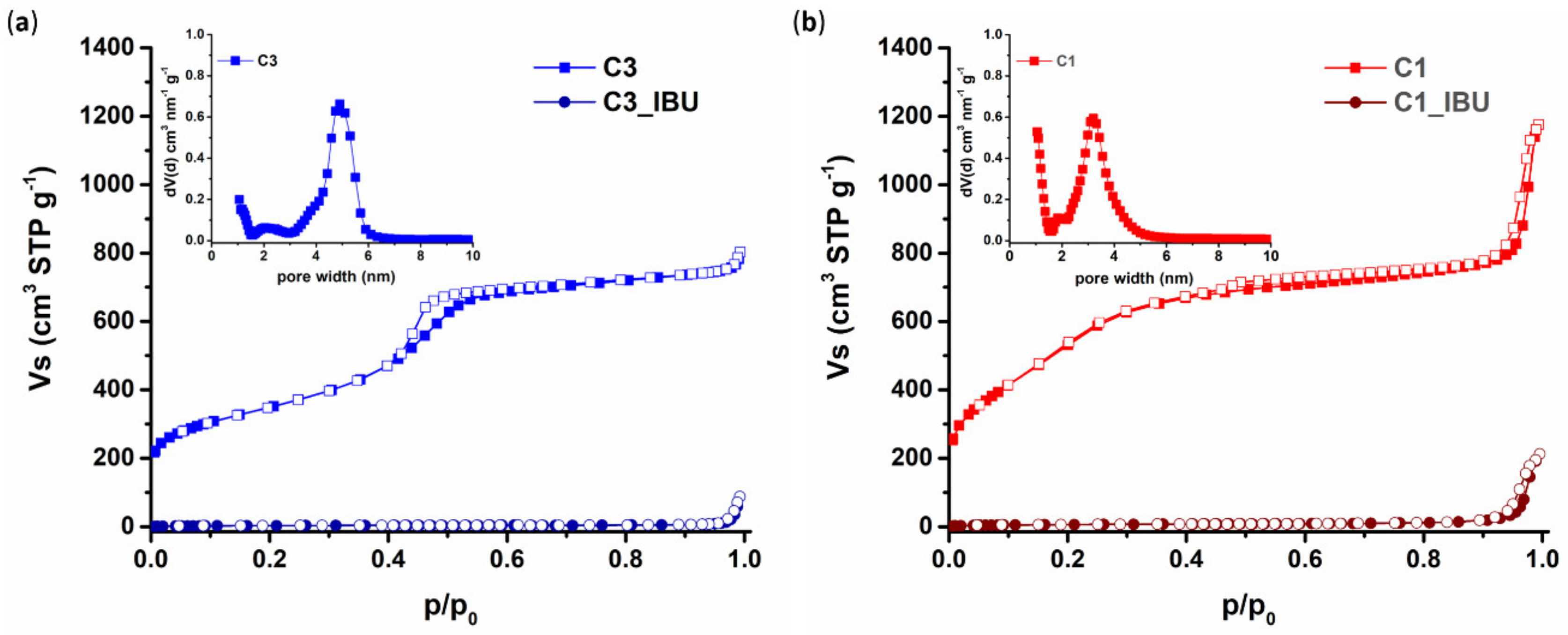
The pore properties of the plain and the IBU-loaded carbons were evaluated by N2 adsorption/desorption measurements at 77 K on a volumetric gas adsorption analyzer (Autosorb-1-MP, Quantachrome Inc., FL, Boynton Beach, USA), using ultra-pure (99.999%) N2. Before analysis, the samples (approximately 30–40 mg) were appropriately outgassed for at least 20 h under high vacuum (10−6 mbar). The Brunauer-Emmett-Teller (BET) area values were calculated on the basis of the BET consistency criteria (ISO 9277:2010). The micropore volumes were assumed to be the QSDFT (Quenched Solid Density Functional Theory)-derived cumulative volumes for pores smaller than 2 nm. The total (micro- and meso-pore) volumes (TPV) were estimated at p/p0 = 0.90, whereas the pore size distributions were deduced by using the N2-carbon QSDFT kernel for slit-cylindrical pores (adsorption model).
2.3. Synthesis of Poly(Ether Urethane)
2.3.1. Materials
The poly(ether urethane) (PEU) used in this work was synthesized using the commercially available triblock copolymer Poloxamer® 407 (poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) PEO-PPO-PEO, 12,600 Da, 70% PEO content) as macrodiol, the aliphatic diisocyanate 1,6-hexamethylene diisocyanate (HDI) and the aliphatic cyclic diol 1,4 cyclohexanedimethanol (CDM) as chain extender. All these three building blocks were purchased from Sigma-Aldrich (Milan, Italy) and used after drying/purification procedures. In detail, P407 was anhydrified under reduced pressure at 100 °C overnight and then equilibrated under vacuum at 40 °C until use, HDI was distilled under vacuum to remove moisture and stabilizers and CDM was stored at RT under reduced pressure in a desiccator until usage. Dibutyltin dilaurate (DBTDL) was also purchased from Sigma-Aldrich (Milan, Italy) and used as received to catalyze the PEU synthesis reaction. Anhydrified 1,2-dichloroethane (DCE) was prepared by storing DCE (Carlo Erba Reagents, Cornaredo, Milan, Italy) over activated (120 °C, atmospheric pressure, overnight) molecular sieves (3 Å, Sigma-Aldrich, Milan, Italy) overnight under inert conditions (i.e., nitrogen flow). All other solvents required for PEU synthesis were purchased from Carlo Erba Reagents (Cornaredo, Milan, Italy) in the analytical grade and used as received. All glassware used for PEU synthesis was stored in an oven at 120 °C until use.
2.3.2. Poly(Ether Urethane) Synthesis Protocol
The poly(ether urethane) used in this work to design hybrid sol-gel systems embedding OMCs was synthesized according to an already published protocol [
43]. Briefly, the synthesis was conducted in two-steps under inert conditions. Initially, a P407 solution in anhydrous DCE was prepared at 20%
w/v concentration and equilibrated at 80 °C. Then, HDI (2:1 molar ratio with respect to P407) and a catalytic amount of DBTDL (0.1%
w/
w with respect to P407) were added to start the prepolymerization step, which lasted 150 min at 80 °C. At the end of the first step, the polymerization system was cooled down to 60 °C and a CDM solution previously prepared in anhydrous DCE (3%
w/v concentration) was added to initiate the chain extension reaction. After 90 min, the system was equilibrated at RT, MeOH was added to passivate potential unreacted isocyanate groups and the synthesized PEU was finally collected by precipitation in petroleum ether at 4:1 volume ratio with respect to the whole DCE volume used during the synthesis. After overnight drying under the fume hood, purification was conducted by precipitating a PEU-concentrated solution prepared in DCE in a mixture of diethyl ether/MeOH (98:2
v/v, 5:1 volume ratio with respect to DCE). The purified PEU was then collected through centrifugation (MIKRO 220R, Hettich, Tuttlingen, Germany, 20 min, 6000 rpm, 0 °C), dried under the fume hood overnight and finally stored under an inert atmosphere until usage.
Hereafter, the synthesized PEU will be referred to with the acronym CHP407, where C, H and P407 refer to CDM, HDI and Poloxamer® 407, respectively.
2.4. Chemical Characterization of as-Synthesized Poly(Ether Urethane)
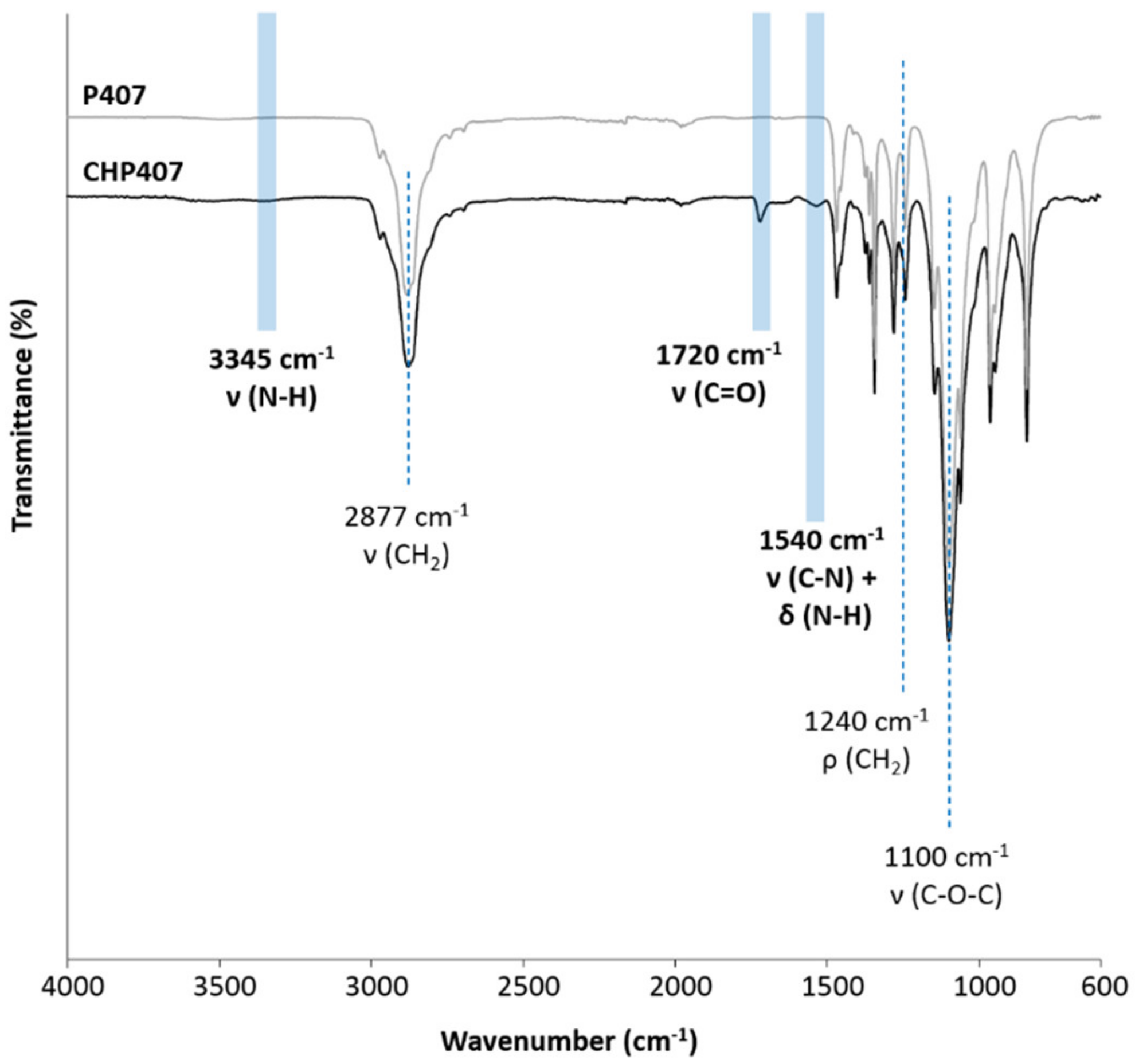
The successful synthesis of CHP407 poly(ether urethane) was first assessed through its chemical characterization by means of Size Exclusion Chromatography (SEC), Proton and Carbon Nuclear Magnetic Resonance (1H and 13C NMR, respectively) spectroscopy and Attenuated Total Reflectance Fourier Transformed Infrared (ATR-FTIR) spectroscopy.
SEC analysis was conducted using an Agilent Technologies 1200 Series (Agilent Technologies Inc., Santa Clara, CA, USA) equipped with a Refractive Index Detector (RID) and two Waters Styragel columns (HR4 and HR1), both equilibrated at 55 °C. A LiBr (Sigma-Aldrich, Milan, Italy) solution in N,N-dimethylformammide (DMF, HPLC grade, Carlo Erba Reagents, Cornaredo, Milan, Italy) (0.1% w/v) was used as eluent for the analysis at a 0.5 mL/min flow rate. The analyzed sample was prepared by filtering the CHP407 solution (2 mg/mL concentration in the eluent phase) with a syringe filter (0.45 μm pore size, polytetrafluoroethylene membrane, Carlo Erba Reagents, Cornaredo, Milan, Italy). As a result of the chromatographic analysis, the RID signal was registered as a function of elution time. Starting from these data and a calibration curve based on poly(methyl methacrylate) standards (Number Average Molecular Weight ranging between 940 and 214,600 Da), the Agilent ChemStation software finally estimated the CHP407 molecular weight distribution profile and its characteristic parameters, namely the Number and Weight Average Molecular Weight values ( and , respectively) and the polydispersity index ().
1H and
13C NMR spectra of synthesized CHP407 were recorded using a Bruker Avance NEO instrument (Bruker Italia S.r.l., Milan, Italy) equipped with a 11.74 T magnet (500 MHz
1H Larmor Frequency) and a Bruker SmartProbe (Bruker Italia S.r.l., Milan, Italy). Analyses were conducted at 300 K on CHP407 samples prepared using d6_DMSO as solvent.
1H and
13C NMR spectra resulted from 32 and 10,000 scans, respectively. Registered spectra were then analyzed using MestReNova software (
https://mestrelab.com/, Mestrelab Research S.L., Santiago de Compostela, Spain) by referring to the residual d6-DMSO proton and carbon signals at 2.5 and 39.5 ppm, respectively, for chemical shift scale.
ATR-FTIR analysis was conducted on both CHP407 and P407 powder using a Perkin Elmer Spectrum 100 instrument (Perkin Elmer, Waltham, MA, USA) equipped with a diamond crystal (UATR KR55, Perkin Elmer, Waltham, MA, USA) ATR set-up. ATR-FTIR spectra of the polymers were registered at RT and resulted from 16 scans recorded between 4000 and 600 cm−1 wavenumber range, with a resolution of 4 cm−1. ATR-FTIR spectra of the samples were then analyzed and their characteristic peaks were identified using Spectrum software (Perkin Elmer, Waltham, MA, USA).
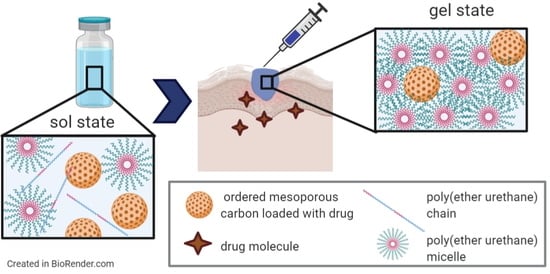
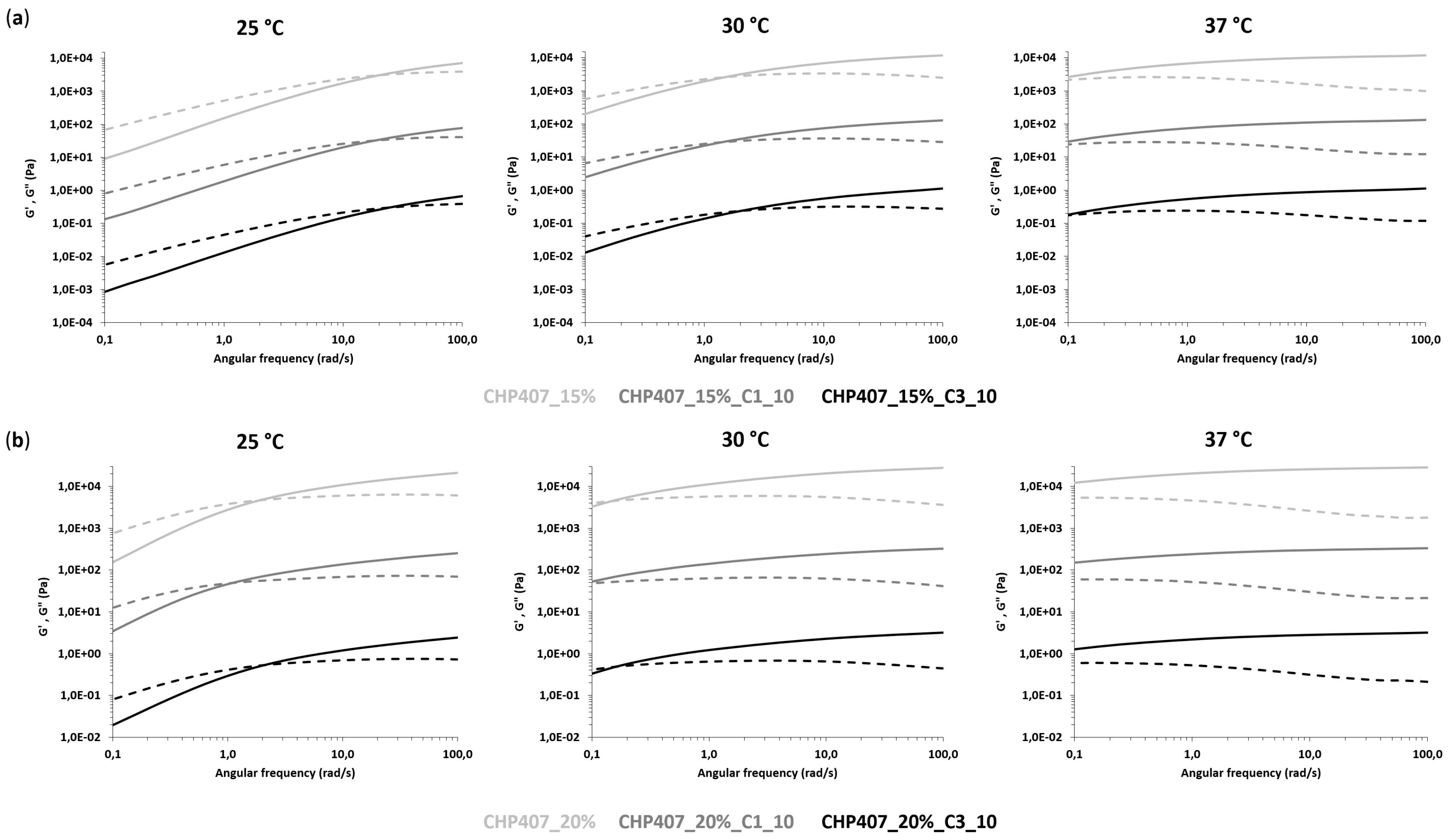
2.5. Design and Characterization of Hybrid Sol-Gel Systems Based on Thermosensitive PEU Hydrogels and OMCs
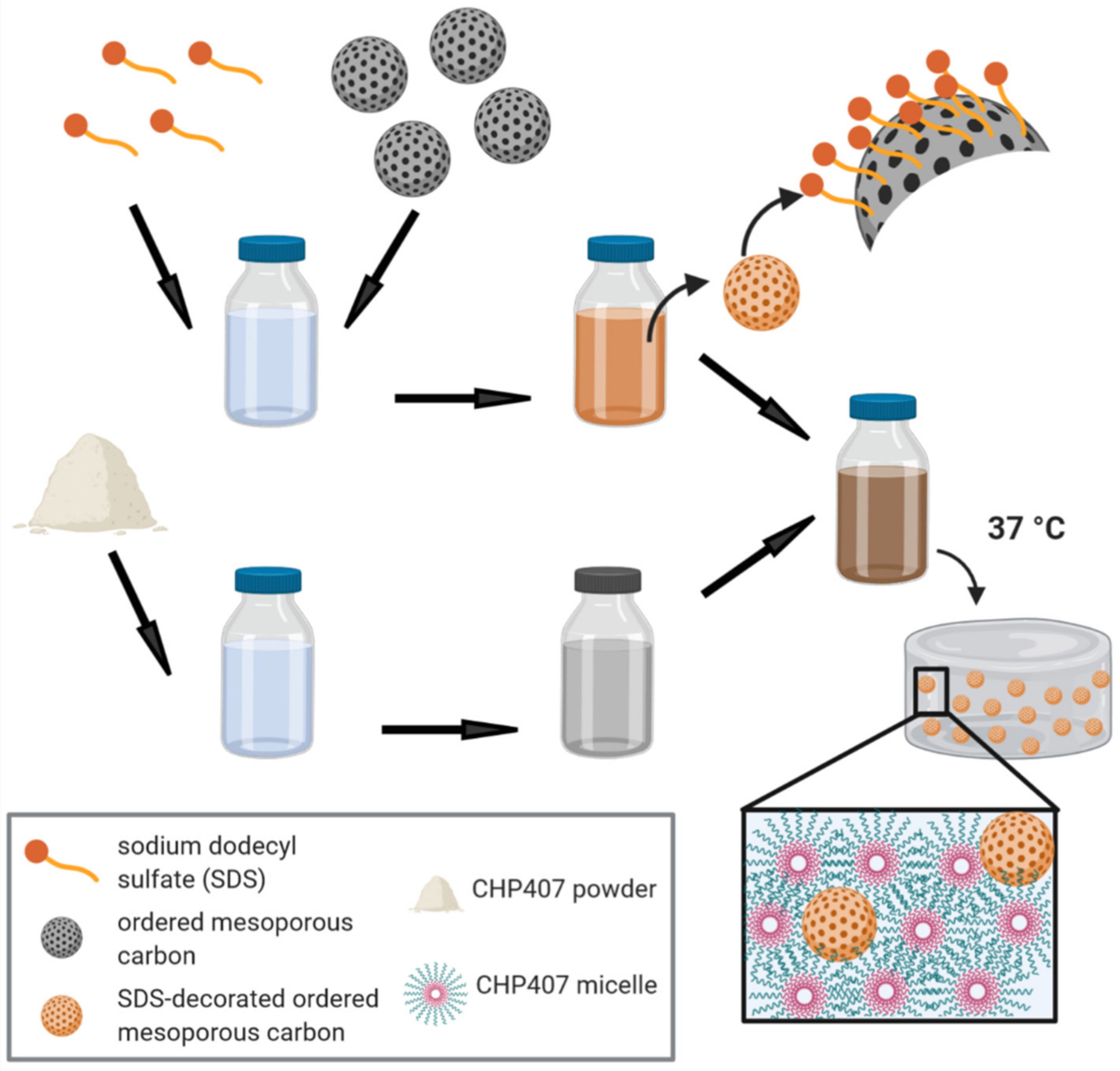
2.5.1. Hydrogel Preparation Protocol
CHP407-based sol-gel systems were prepared at a final polymer concentration of 15 and 20%
w/v. These compositions were selected based on previous works where we reported the complete sol-to-gel transition curve of thermosensitive hydrogels based on PEUs with chemical composition similar to CHP407 [
44,
45]. Indeed, formulations at 15 and 20%
w/v PEU concentration have been reported to exhibit fast gelation in physiological conditions, injectability and prolonged stability in aqueous environments. Hybrid PEU/OMC formulations were prepared with a similar approach to the one we have already described [
35,
36,
37], i.e., by mixing a PEU aqueous solution with an OMC aqueous dispersion to achieve a final PEU concentration of 15 or 20%
w/v and an OMC content of 10 mg/mL. Briefly, the required amount of CHP407 powder was weighed and dissolved at 4 °C overnight in a volume of physiological solution (0.9% NaCl) corresponding to 90% of the total volume required to solubilize it at a 15 or 20%
w/v concentration. Then, the required amount of each OMC was dispersed in a volume of double demineralized water containing sodium dodecyl sulfate (SDS, 1%
w/v, Sigma-Aldrich, Milan, Italy) corresponding to 10% of the total volume required to achieve a PEU concentration of 15 or 20%
w/v in the final hybrid formulations. OMC dispersion was then added to CHP407 samples kept in the sol state in a water bath and the resulting formulations were mixed with a vortex to homogeneously disperse the particles. For instance, in a typical experimental set-up, 150 mg of CHP407 were solubilized in 900 µL of physiological solution. Then, 10 mg of OMCs were weighed and dispersed in an SDS aqueous solution (100 µL, 1%
w/v), which was then mixed with CHP407 solution to obtain a hybrid sol-gel system with CHP407 and OMCs at 15%
w/v and 10 mg/mL concentration, respectively. Similar samples embedding OMCs previously loaded with IBU (OMC_IBU) were also prepared.
For comparison, pure CHP407 sol-gel systems were prepared at 15 and 20% w/v concentration in an aqueous medium consisting of a mixture of physiological solution and SDS solution (1% w/v in double demineralized water) at 90:10 volume ratio.
Table 1 summarizes the compositions and acronyms of the formulations designed and characterized in this work.
2.5.2. Hybrid Hydrogel Characterization
The capability of CHP407-based hybrid formulations to undergo a temperature-driven sol-to-gel transition was assessed through tube inverting test and rheological characterization.
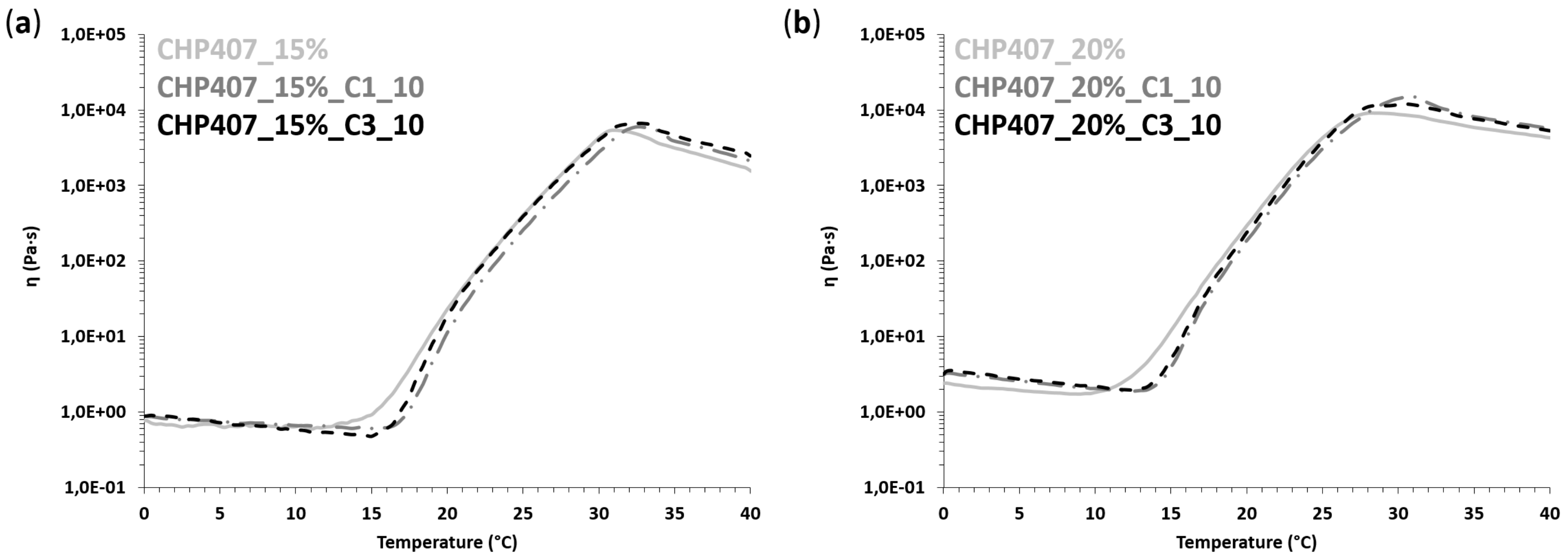
A tube inverting test was performed to estimate the gelation temperature (i.e., the Lower Critical Gelation Temperature, LCGT) of the developed sol-gel systems. To this aim, all the investigated formulations (1 mL) were prepared in Bijou sample containers (Thermo Scientific™ Sterilin™, Waltham, MA, USA ) with 17 mm internal diameter to avoid results dependence over sample geometry and volume. Samples were prepared according to the previously described protocol and subjected to a controlled temperature increase from 4 to 40 °C, at 1 °C/step; at each step the temperature was kept stable for 5 min and was followed by vial inversion and visual inspection for 30 s. Conditions of sol and gel were defined based on the presence of a flow along vial walls during sample inversion. The temperature characterized by the complete absence of flow during the 30 s of vial inversion was defined as the LCGT of the formulation. A tube inverting test was also conducted at a constant temperature of 37 °C to estimate the time required by the samples to undergo complete gelation at physiological temperature. To this aim, samples were incubated at 37 °C and the vials were inverted every 1 min to assess the presence or absence of sample flow along their walls. The incubation time at which no sample flow was detected defined the time required for the investigated formulation to undergo complete sol-to-gel transition at 37 °C.
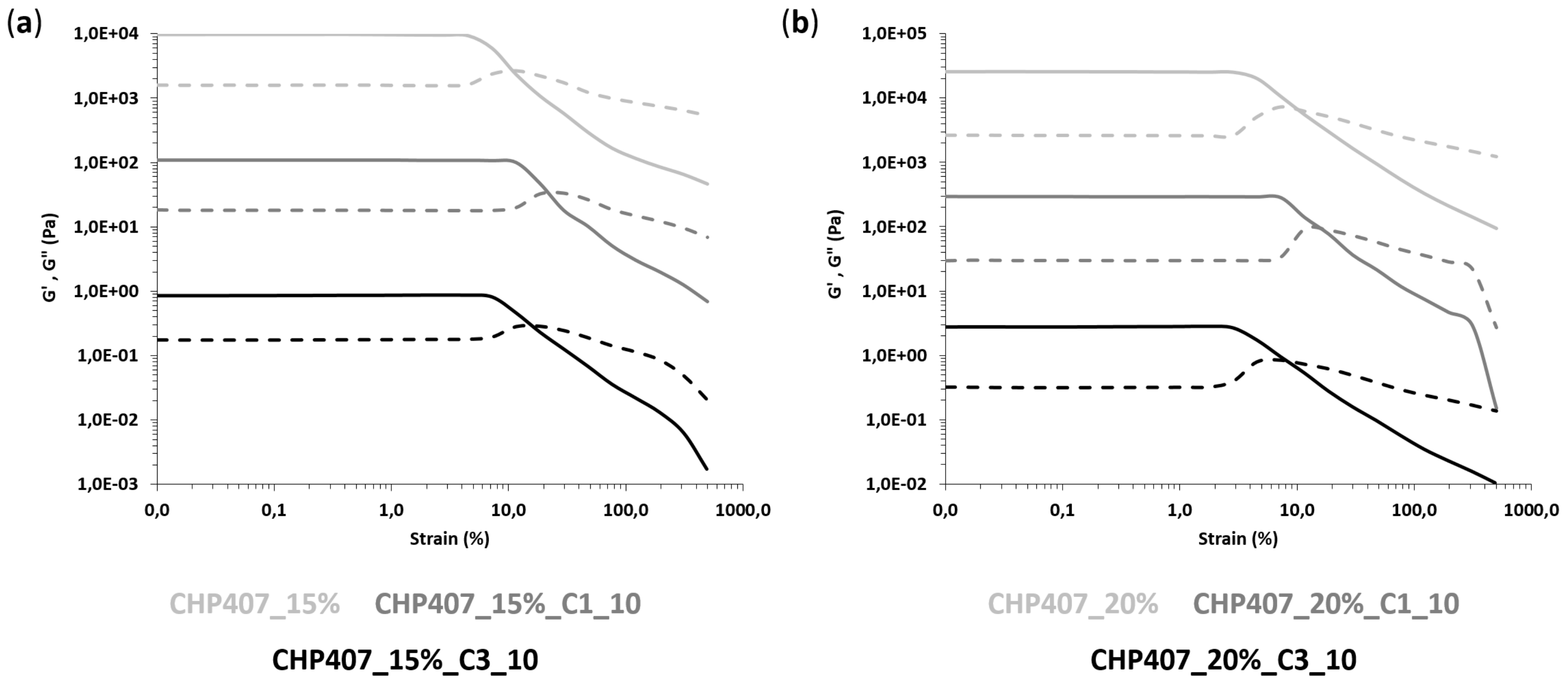
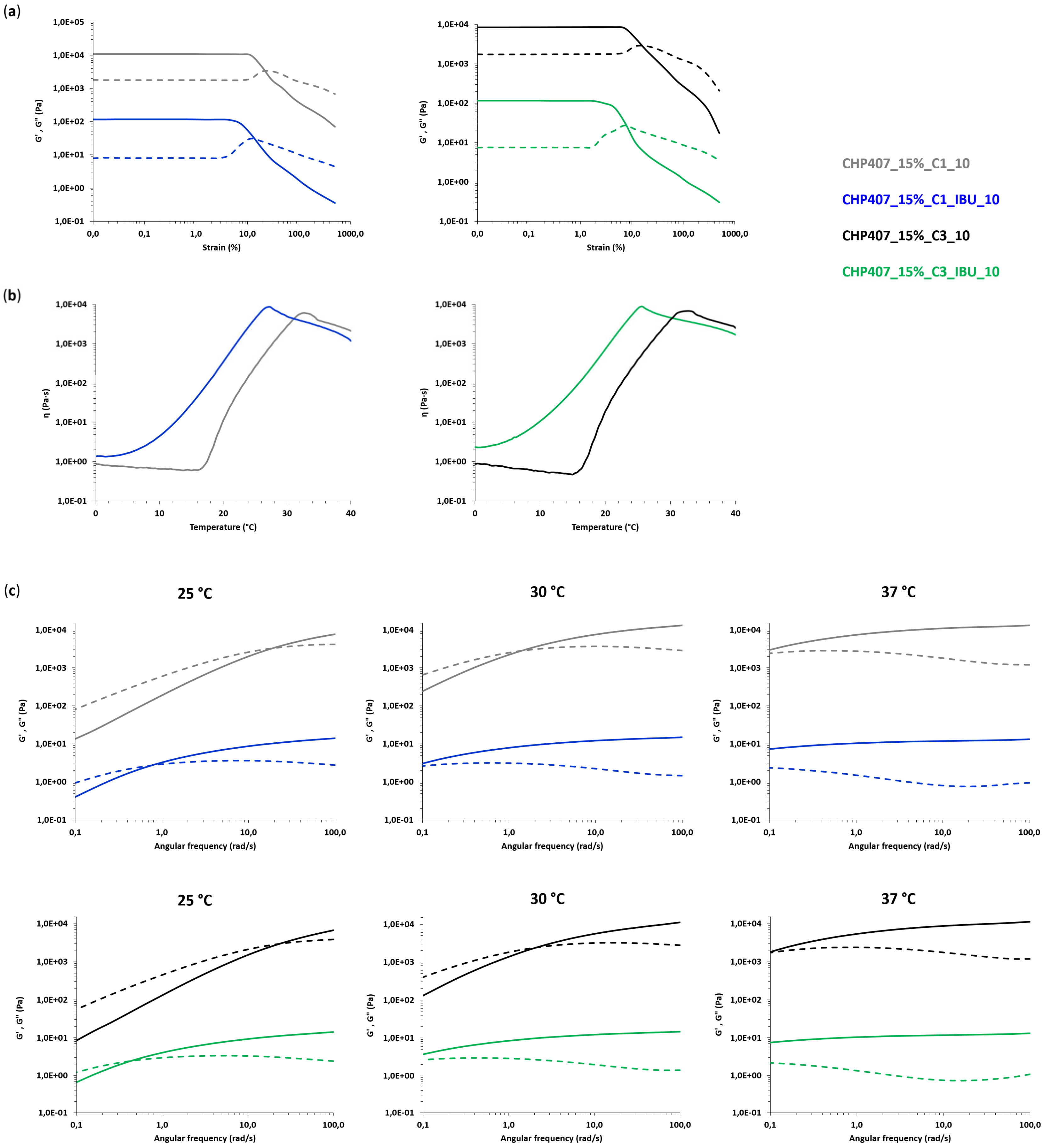
CHP407-based formulations were also characterized through rheological analyses (MCR302, Anton Paar GmbH, Graz, Austria, parallel plate geometry 50 mm, Peltier system for temperature control). First, strain sweep tests were performed at 37 °C and 10 Hz within the strain range from 0.01 to 500% to characterize the gels’ capability to withstand an applied deformation, and define their linear viscoelastic (LVE) region (i.e., the strain range characterized by a constant value of the measured storage modulus) and Yield Stress (YS, i.e., the value of shear stress at the maximum of loss modulus). The progressive gel formation with an increasing temperature was monitored through temperature ramp tests within the temperature range from 0 to 40 °C, at 2 °C/min and 0.1 Hz. In parallel, the evolution of the sample network from the sol to the gel phase, encompassing the biphasic state, was studied through frequency sweep tests, which were carried out at three different temperatures (i.e., 25, 30 and 37 °C) and strain within the previously defined LVE region, within the angular frequency range from 0.1 to 100 rad/s. Samples were loaded on the lower plate of the instrument equilibrated at 0 °C, then the gap was fixed at 0.6 mm and the system was heated at the testing temperature and left to equilibrate for 10 min before the beginning of the analysis (for temperature ramp tests, equilibration was performed at 0 °C).
2.6. Ibuprofen Release from Hybrid Sol-Gel Systems
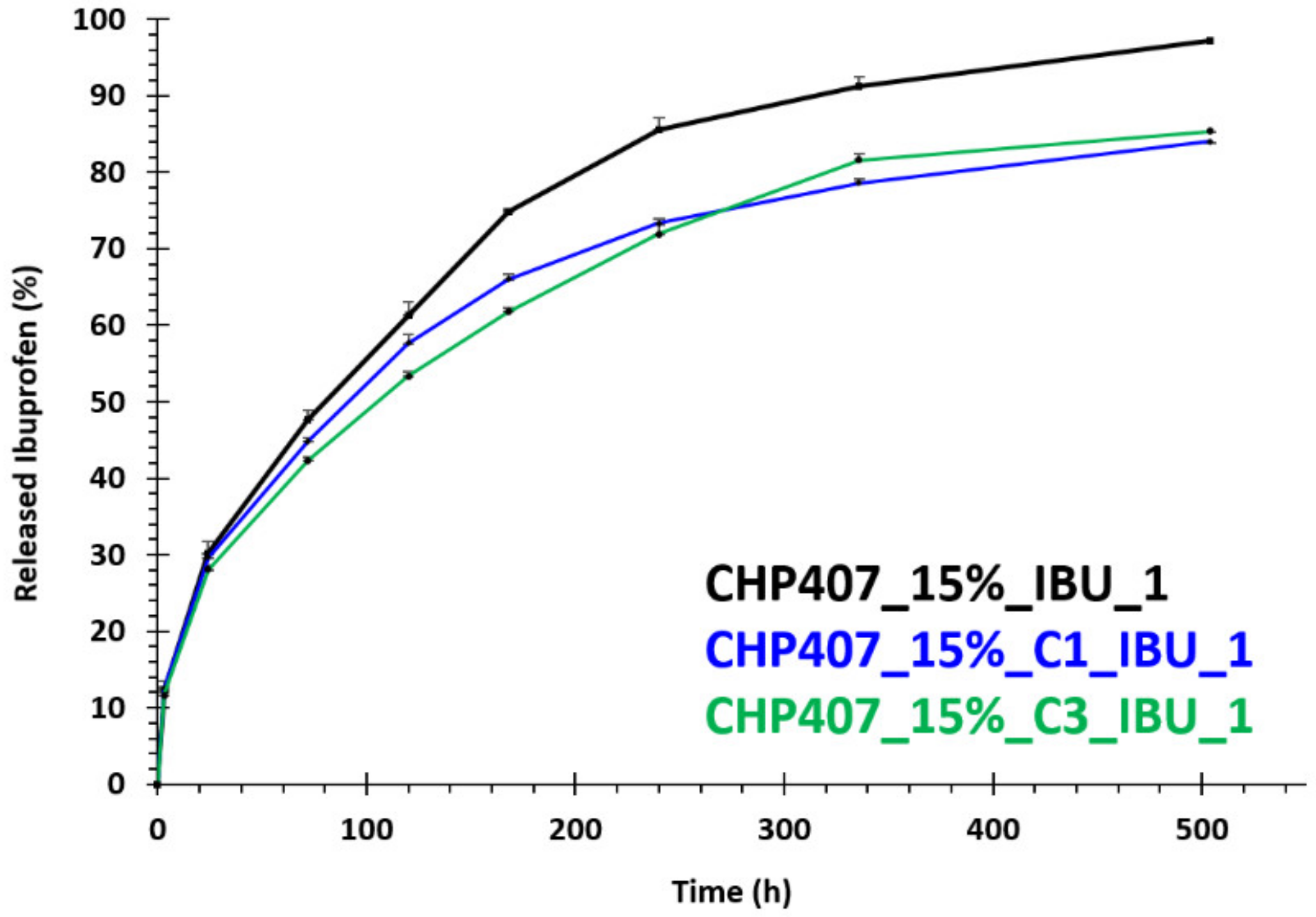
In order to assess the capability of CHP407/OMC hybrid formulations to work as a reservoir of drugs (e.g., ibuprofen) and progressively release it in the surrounding aqueous environment, IBU release tests were conducted in physiological-like conditions, i.e., at 37 °C and using Phosphate Buffered Saline (PBS, pH 7.4) as a releasing medium. CHP407/OMC hybrid samples (1 mL) were prepared according to the previously described protocol by embedding IBU-loaded OMCs (C1_IBU and C3_IBU) at a final carbon concentration of 1 mg/mL (samples CHP407_15%_C1_IBU_1 and CHP407_15%_C3_IBU_1). For comparison purposes, CHP407-based hydrogels (1 mL) with the same polymer content and loaded with IBU as such at the same concentration as that of CHP407/OMC hybrid samples were prepared according to Boffito and Pontremoli et al. [
35] (CHP407_15%_IBU).
All investigated formulations were first incubated at 37 °C in an incubator (LabTech, Sorisole, Italy) for 15 min to allow their complete gelation; then 1 mL of PBS (kept at 37 °C in the incubator to equilibrate) was added to each sample and the release tests started. The release medium was then collected and completely refreshed at 3 h, 24 h, 3 d, 5 d, 7 d, 10 d, 14 d and 21 d incubation times. Collected release media were analyzed through spectrophotometric analysis (UV-1800 spectrophotometer, Shimadzu Corporation, Kyoto, Japan) within the spectral range 400–190 nm by analyzing the IBU absorbance peak at 222 nm. Released ibuprofen was finally quantified by referring to a calibration curve based on IBU standard samples prepared by solubilizing the drug in PBS at concentrations within the range 2.5–200 µg/mL.
2.7. Statistical Analysis
Ibuprofen release tests were conducted in triplicate and results are reported as mean ± standard deviation. GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA, version 5.03, 2009;
http://www.graphpad.com) was employed to subject ibuprofen release data to a Two-way ANOVA analysis followed by Bonferroni’s multiple comparison test.
4. Conclusions
The design of drug delivery systems that carry a huge amount of active molecules and release them to a target biological compartment according to a controlled and sustained profile represents an overwhelming advancement in the biomedical field. Indeed, such an approach overcomes the main drawbacks of traditional drug administration routes (e.g., oral and systemic administration), limiting the need for multiple administrations and opening the way to the possibility to set up personalized therapies. With this perspective in mind, in this work we reported on the possibility to design hybrid injectable thermosensitive sol-gel formulations. To this end, we combined ordered mesoporous carbons and thermosensitive poly(ether urethane)-based sol-gel systems, relying on the well-known high drug loading capability, biocompatibility and chemical stability of the former [
20] and the improved gelation, mechanical properties and residence time of the latter compared to commercial P407-based hydrogels [
44]. OMCs with rod-like and spherical shapes were successfully synthesized and completely filled with ibuprofen (100% loading yield, 1:1 carbon/IBU weight ratio). Meanwhile, a high molecular weight poly(ether urethane) (
72 kDa, D 1.7) was obtained starting from P407, HDI and CDM as building blocks. The infiltration of ibuprofen in the pores of the OMCs was first assessed through DSC analysis, which evidenced the absence of the crystalline drug for both particle types. The process of OMC loading into the hydrogel vehicle phase did not negatively affect the gelation potential of the investigated formulations. Interestingly, the gelation process of the hydrogels turned out to be influenced by the OMC surface decoration with the surfactant SDS. Specifically, its hydrophilic heads probably took an active part in the sol-to-gel transition through the formation of hydrogen bonds with CHP407 chains, thus allowing OMCs to act as nodes within the gel network. In addition, OMC geometrical features also appeared to affect gel properties, with smaller spherical C1 particles better integrating within CHP407 gels. The high yield of IBU encapsulation into OMCs allowed the loading of a high drug content into the hydrogels through the embedding of a small amount of OMCs. Furthermore, OMCs turned out to effectively act as drug reservoirs, enabling a prolonged and sustained release of their cargo over time. Indeed, while hydrogels containing ibuprofen as such completely released it within 3 weeks, OMC-loaded systems delivered ca. the 85% of their payload within the same time interval. Such features make the formulations developed in this work able to perfectly address the previously mentioned demands, thus opening the way for their potential exploitation in the biomedical field to treat a wide range of pathological states (e.g., chronic skin wounds, delayed bone healing, cancer, damaged cartilage) upon a proper selection of the therapeutic agents to load. However, in their current formulation, the here-characterized hybrid sol-gel systems would not be suitable for application in photothermal therapy due to the LCGT behavior of CHP407 poly(ether urethane)-based aqueous solutions. To make our platform suitable for this application, PEU LEGO-like structure could be exploited to include in the polymer backbone proper functional groups (e.g., thiols, amines, aldehyde, carboxyl groups), which will allow a secondary stabilization of the thermo-induced gel network through the formation of chemical crosslinks making the resulting system unresponsive to further temperature changes. Finally, by exploiting both the micellar nature of the hydrogels and the mesoporous framework of OMCs, formulations releasing a cocktail of compounds, each according to a specific time schedule, could be developed for advanced therapeutic applications.

 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}