Coupling Hydrogenation of Guaiacol with In Situ Hydrogen Production by Glycerol Aqueous Reforming over Ni/Al2O3 and Ni-X/Al2O3 (X = Cu, Mo, P) Catalysts

 , ,
, ,
Abstract
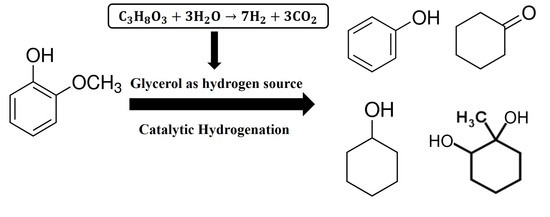
:
1. Introduction
2. Experimental
2.1. Reagents
2.2. Catalyst Preparation
2.3. Catalyst Characterization
2.4. Catalytic Activity in the Hydrogenation of Guaiacol with In-Situ Glycerol Reforming as a Hydrogen Source
2.5. Gas Product Determination
2.6. Liquid Product Determination
2.7. Solid Product Determination
3. Results and Discussion
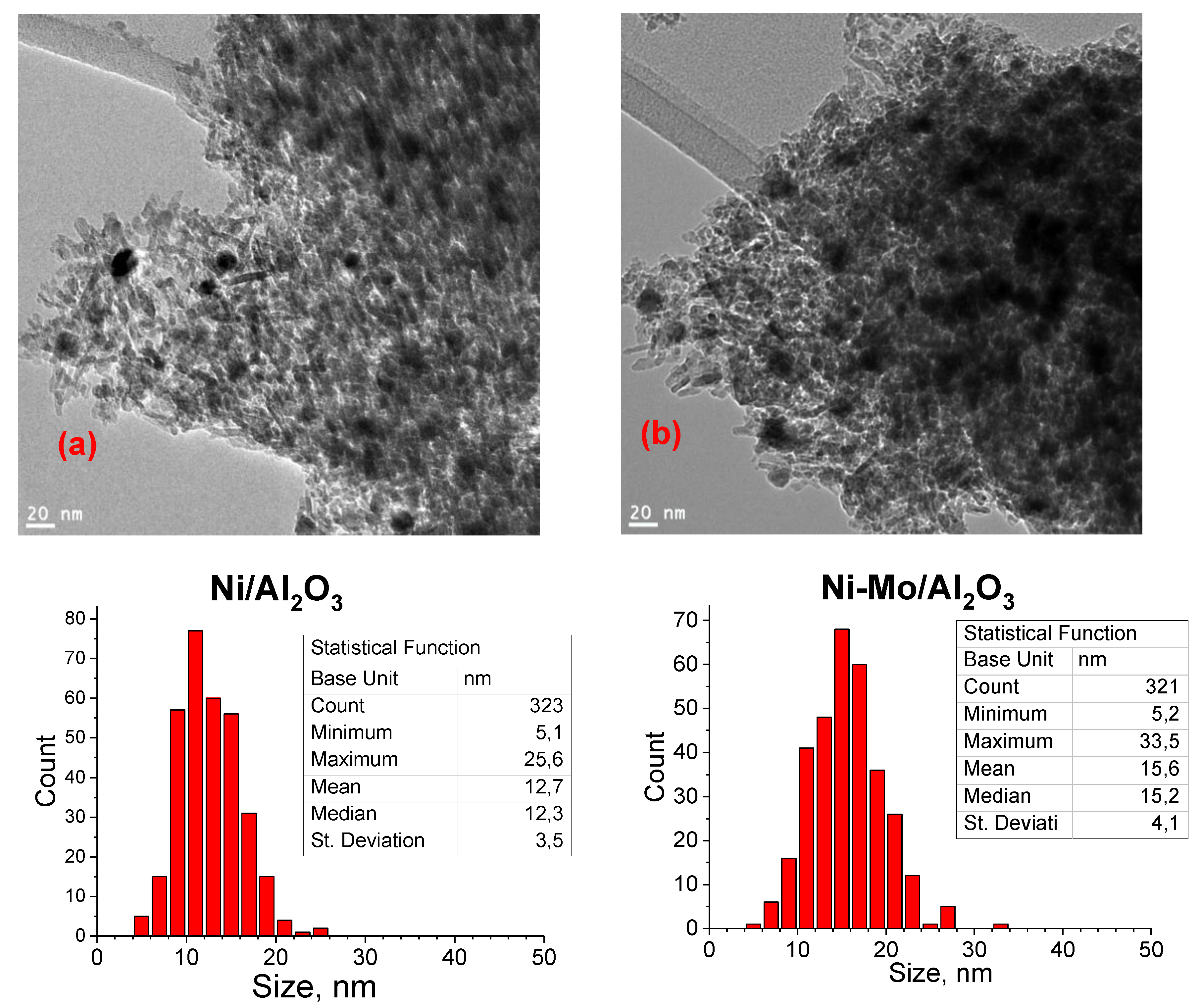
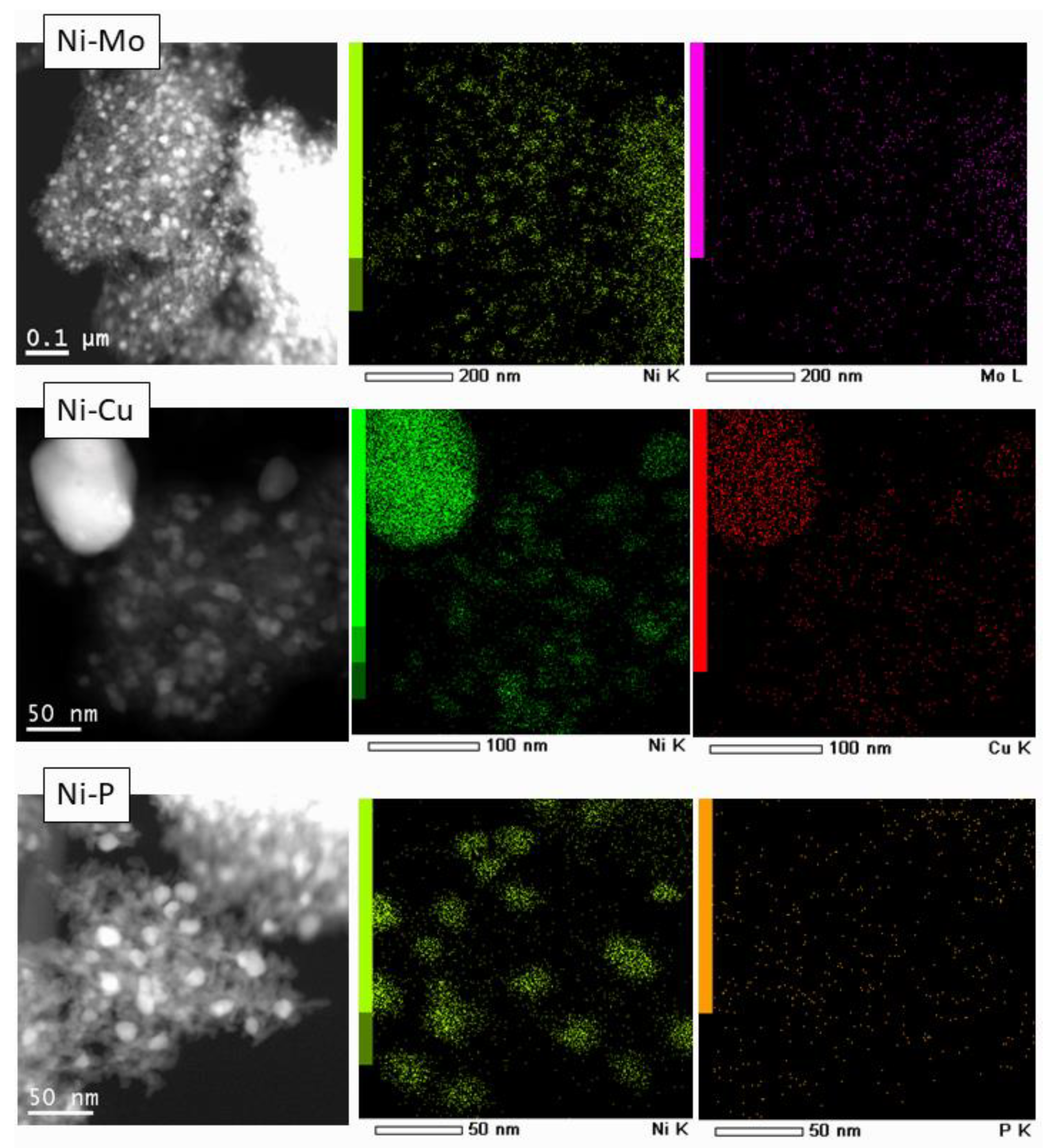
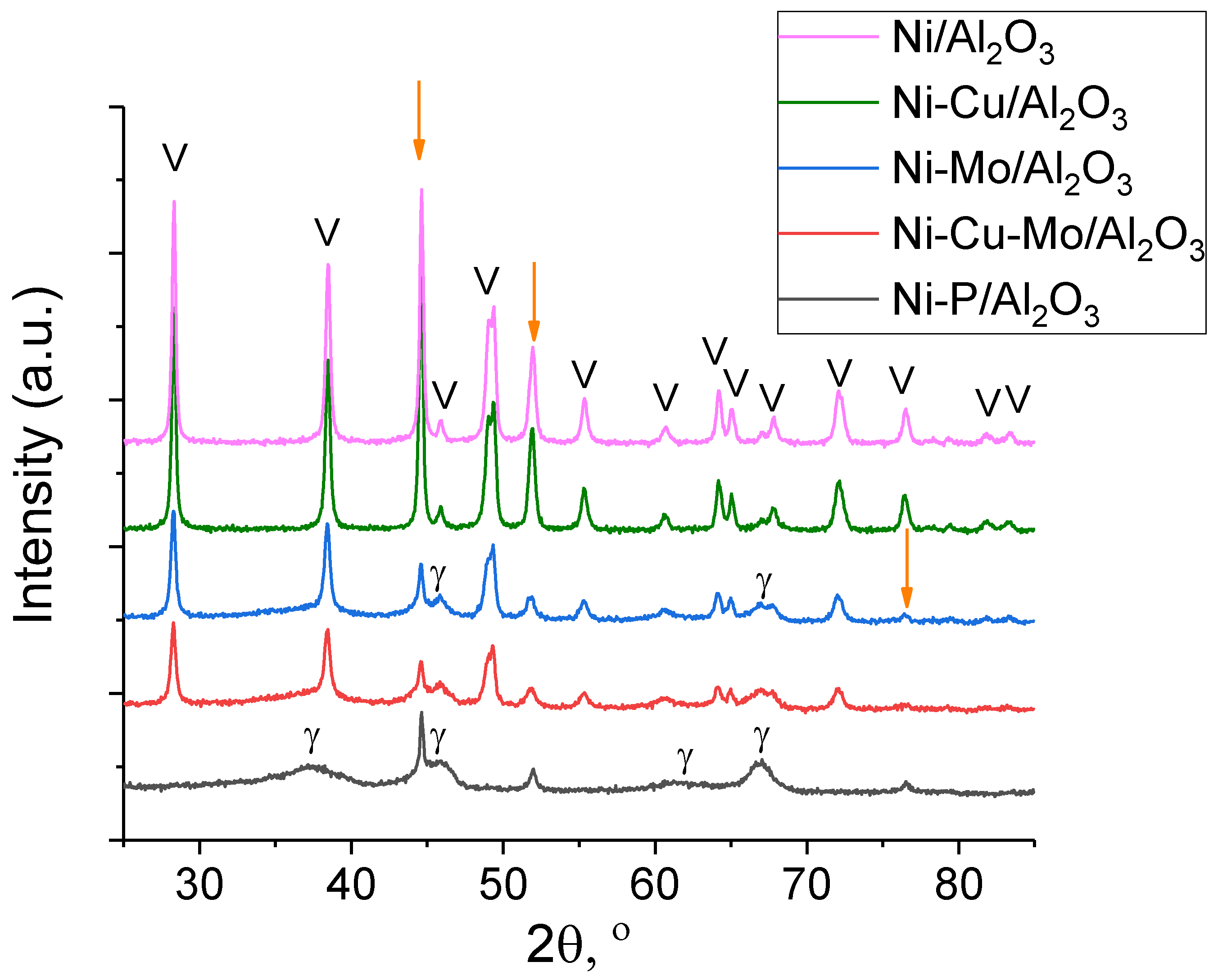
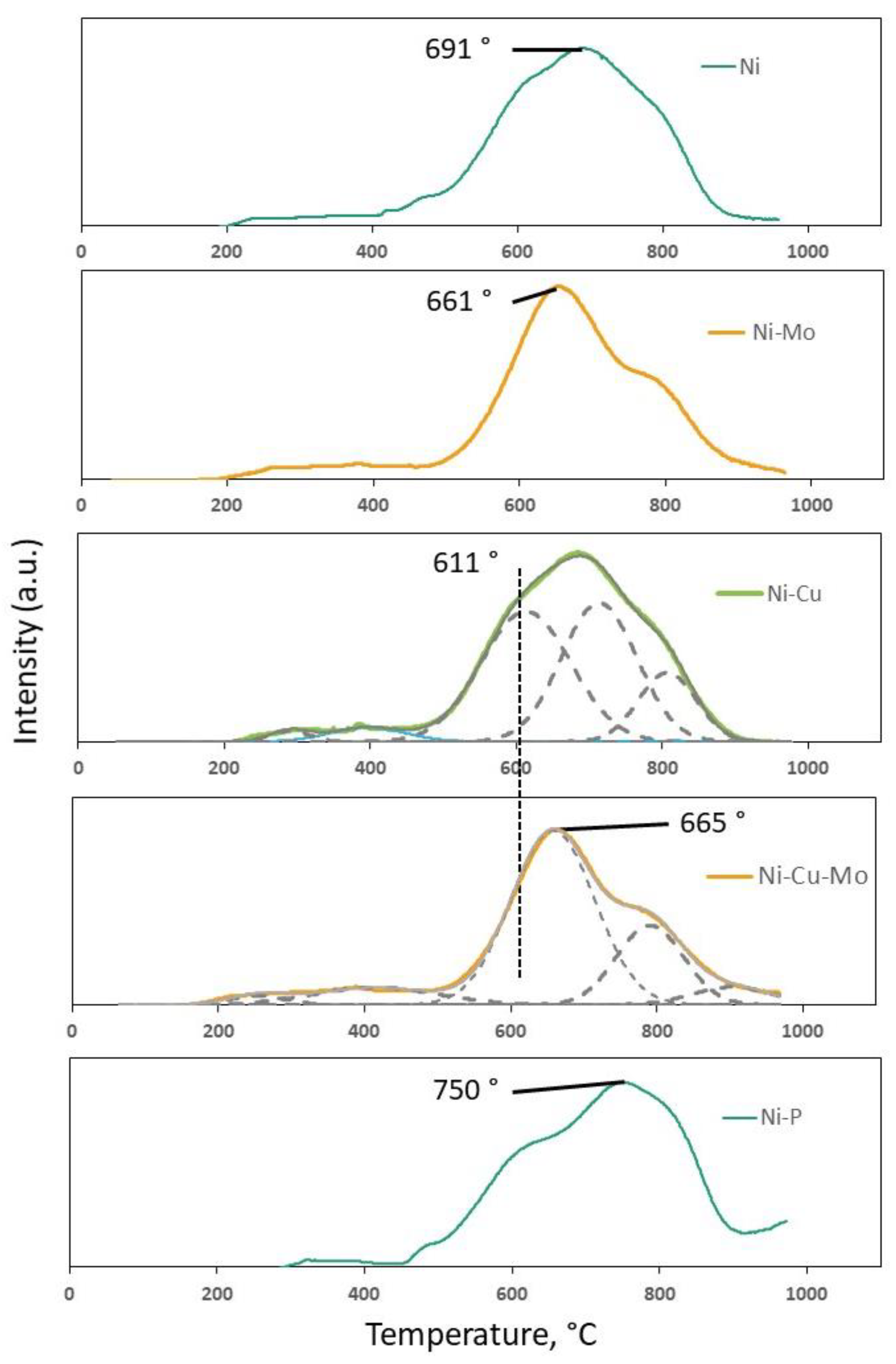
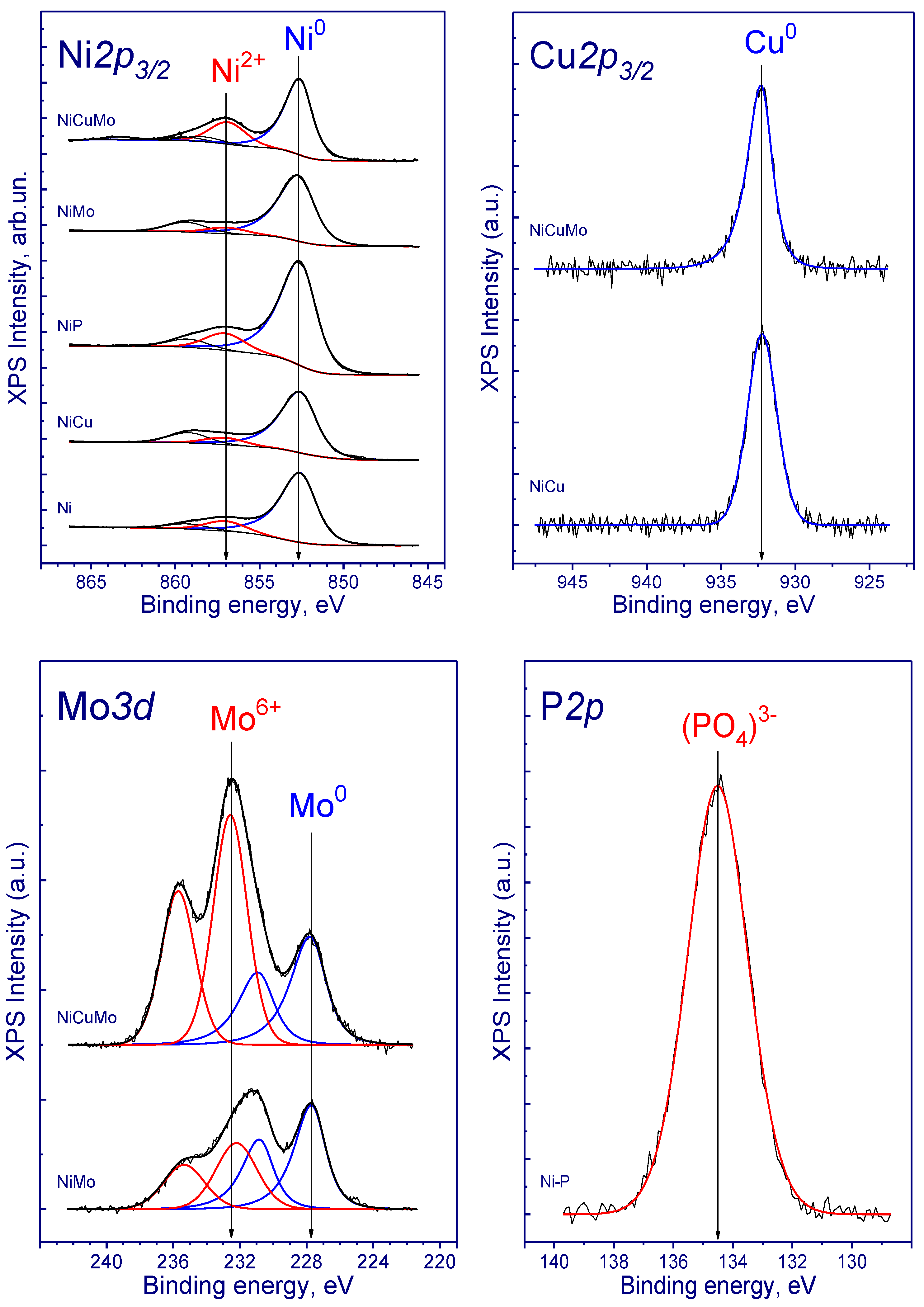
3.1. Catalyst Characterization
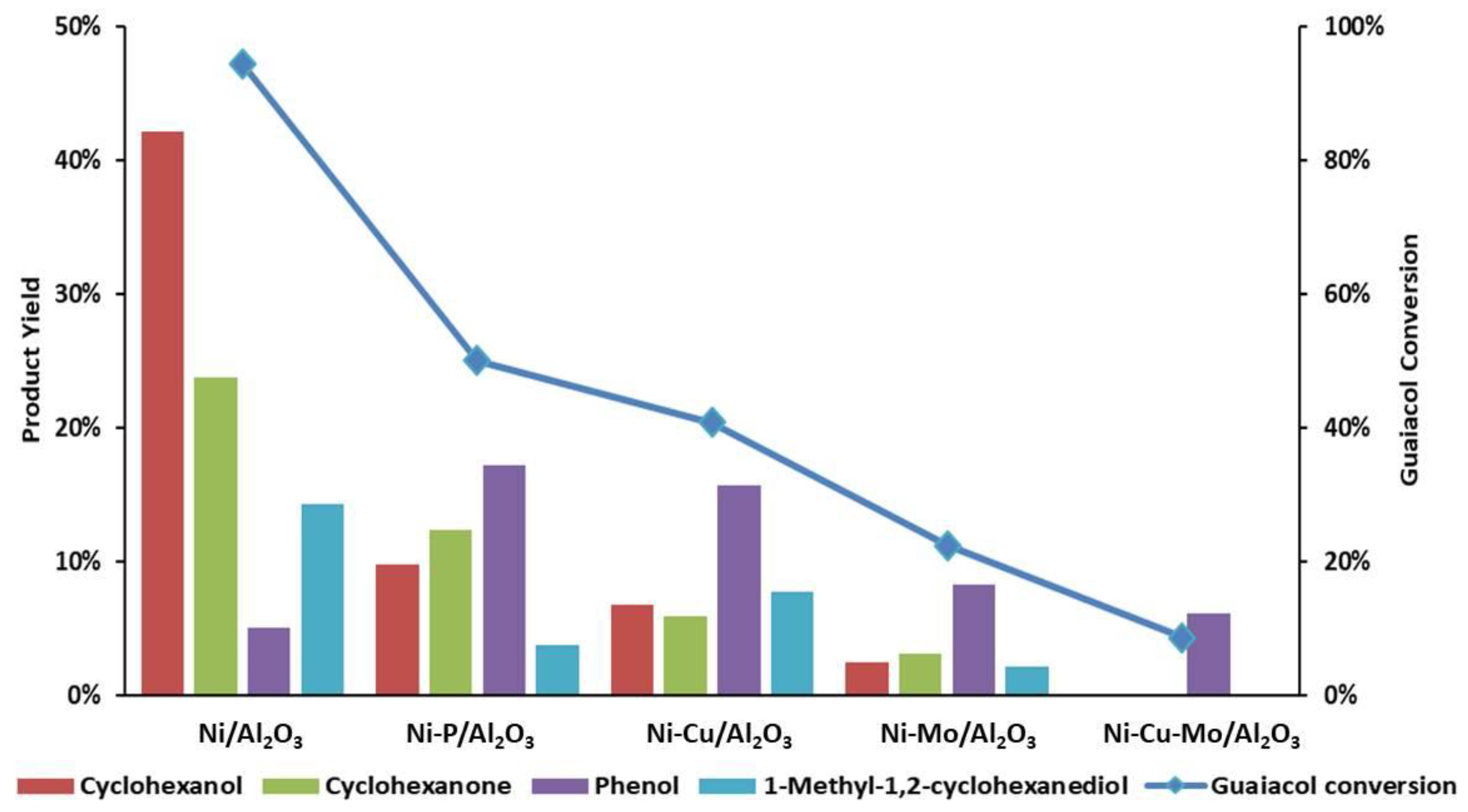
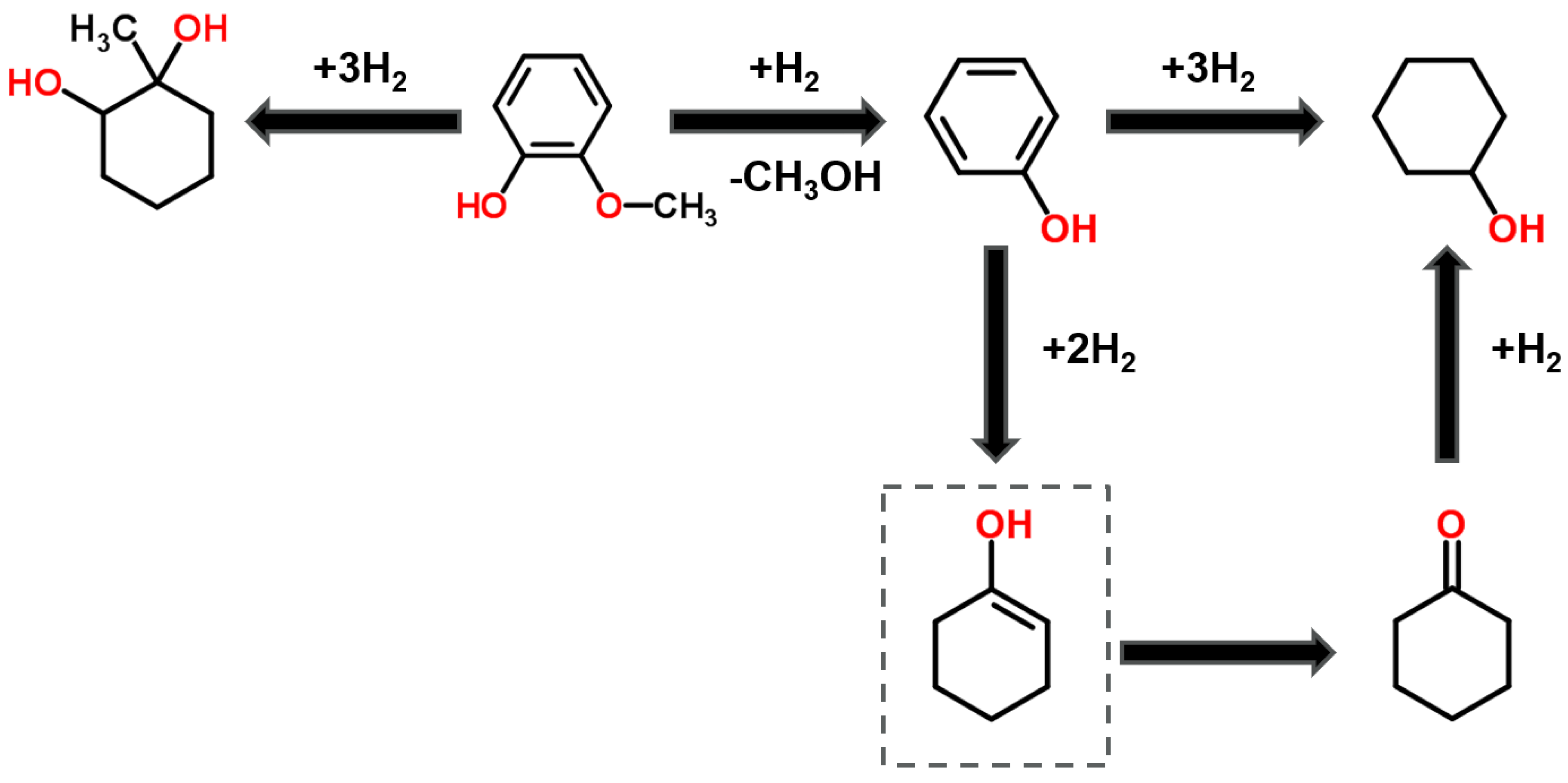
3.2. In-Situ Hydrogenation of Guaiacol with the Ni-Based Alumina-Supported Catalysts
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, D.; Pittman Jr, C.; Steele, P. Pyrolysis of wood/biomass for bio-oil: A critical review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Wang, H.; Male, J.; Wang, Y. Recent advances in hydrotreating of pyrolysis bio-oil and its oxygen-containing model compounds. Acs Catal. 2013, 3, 1047–1070. [Google Scholar] [CrossRef]
- Yakovlev, V.A.; Khromova, S.A.; Sherstyuk, O.V.; Dundich, V.O.; Ermakov, D.Y.; Novopashina, V.M.; Lebedev, M.Y.; Bulavchenko, O.; Parmon, V.N. Development of new catalytic systems for upgraded bio-fuels production from bio-crude-oil and biodiesel. Catal. Today 2009, 144, 362–366. [Google Scholar] [CrossRef]
- Arcelus-Arrillaga, P.; Pinilla, J.L.; Hellgardt, K.; Millan, M. Application of water in hydrothermal conditions for upgrading heavy oils: A review. Energy Fuels 2017, 31, 4571–4587. [Google Scholar] [CrossRef]
- Zakzeski, J.; Weckhuysen, B.M.J.C. Lignin solubilization and aqueous phase reforming for the production of aromatic chemicals and hydrogen. ChemSusChem 2011, 4, 369–378. [Google Scholar] [CrossRef]
- Zakzeski, J.; Jongerius, A.; Bruijnincx, P.; Weckhuysen, B. Catalytic lignin valorization process for the production of aromatic chemicals and hydrogen. ChemSusChem 2012, 5, 1602–1609. [Google Scholar] [CrossRef]
- Fisk, C.A.; Morgan, T.; Ji, Y.; Crocker, M.; Crofcheck, C.; Lewis, S.A. Bio-oil upgrading over platinum catalysts using in situ generated hydrogen. Appl. Catal. A Gen. 2009, 358, 150–156. [Google Scholar] [CrossRef]
- Tan, Z.; Xu, X.; Liu, Y.; Zhang, C.; Zhai, Y.; Li, Y.; Zhang, R. Upgrading bio-oil model compounds phenol and furfural with in situ generated hydrogen. Environ. Prog. Sustain. Energy 2014, 33, 751–755. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Li, C.-L.; Wan, H.-P.; Lee, H.-T.; Liu, C.-F. Catalytic hydrodeoxygenation of guaiacol on Rh-based and sulfided CoMo and NiMo catalysts. Energy Fuels 2011, 25, 890–896. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Yagi, T.; Shinobara, S.; Fukunaga, T.; Nakasaka, Y.; Tago, T.; Masuda, T. Production of phenols from lignin via depolymerization and catalytic cracking. Fuel Process. Technol. 2013, 108, 69–75. [Google Scholar] [CrossRef]
- Feng, J.; Yang, Z.; Hse, C.-Y.; Su, Q.; Wang, K.; Jiang, J.; Xu, J. In situ catalytic hydrogenation of model compounds and biomass-derived phenolic compounds for bio-oil upgrading. Renew. Energy 2017, 105, 140–148. [Google Scholar] [CrossRef]
- Kim, M.; Ha, J.-M.; Lee, K.-Y.; Jae, J. Catalytic transfer hydrogenation/hydrogenolysis of guaiacol to cyclohexane over bimetallic RuRe/C catalysts. Catal. Commun. 2016, 86, 113–118. [Google Scholar] [CrossRef]
- Quispe, C.A.; Coronado, C.J.R.; Carvalho, J.A., Jr. Glycerol: Production, consumption, prices, characterization and new trends in combustion. Renew. Sustain. Energy Rev. 2013, 27, 475–493. [Google Scholar] [CrossRef]
- Torres, D.; Arcelus-Arrillaga, P.; Millan, M.; Pinilla, J.L.; Suelves, I. Enhanced reduction of few-layer graphene oxide via supercritical water gasification of glycerol. Nanomaterials 2017, 7, 447. [Google Scholar] [CrossRef] [Green Version]
- Putra, R.D.D.; Trajano, H.L.; Liu, S.; Lee, H.; Smith, K.; Kim, C.S. In-situ glycerol aqueous phase reforming and phenol hydrogenation over Raney Ni®. Chem. Eng. J. 2018, 350, 181–191. [Google Scholar] [CrossRef]
- Yin, W.; Venderbosch, R.H.; Alekseeva, M.V.; Figueirêdo, M.B.; Heeres, H.; Khromova, S.A.; Yakovlev, V.A.; Cannilla, C.; Bonura, G.; Frusteri, F. Hydrotreatment of the carbohydrate-rich fraction of pyrolysis liquids using bimetallic Ni based catalyst: Catalyst activity and product property relations. Fuel Process. Technol. 2018, 169, 258–268. [Google Scholar] [CrossRef]
- Alekseeva, M.V.; Rekhtina, M.A.; Lebedev, M.Y.; Zavarukhin, S.G.; Kaichev, V.V.; Venderbosch, R.H.; Yakovlev, V.A. Hydrotreatment of 2-Methoxyphenol over High Ni-Loaded Sol-Gel Catalysts: The Influence of Mo on Catalyst Activity and Reaction Pathways. ChemistrySelect 2018, 3, 5153–5164. [Google Scholar] [CrossRef]
- Yin, W.; Venderbosch, R.H.; Yakovlev, V.A.; Heeres, H.J. Catalytic Hydrotreatment of the Pyrolytic Sugar and Pyrolytic Lignin Fractions of Fast Pyrolysis Liquids Using Nickel Based Catalysts. Energies 2020, 13, 285. [Google Scholar] [CrossRef] [Green Version]
- Kukushkin, R.G.; Bulavchenko, O.A.; Kaichev, V.V.; Yakovlev, V.A. Influence of Mo on catalytic activity of Ni-based catalysts in hydrodeoxygenation of esters. Appl. Catal. B Environ. 2015, 163, 531–538. [Google Scholar] [CrossRef]
- Kukushkin, R.G.; Reshetnikov, S.I.; Zavarukhin, S.G.; Eletskii, P.M.; Yakovlev, V.A. Kinetics of the Hydrodeoxygenation of Ethyl Ester of Decanoic Acid over the Ni–Cu–Mo/Al2O3 Catalyst. Catal. Ind. 2019, 11, 191–197. [Google Scholar] [CrossRef]
- Kukushkin, R.G.; Eletskii, P.M.; Bulavchenko, O.A.; Saraev, A.A.; Yakovlev, V.A. Studying the Effect of Promotion with Copper on the Activity of the Ni/Al2O3 Catalyst in the Process of Ester Hydrotreatment. Catal. Ind. 2019, 11, 198–207. [Google Scholar] [CrossRef]
- Bykova, M.; Ermakov, D.Y.; Khromova, S.A.; Smirnov, A.A.; Lebedev, M.Y.; Yakovlev, V.A. Stabilized Ni-based catalysts for bio-oil hydrotreatment: Reactivity studies using guaiacol. Catal. Today 2014, 220, 21–31. [Google Scholar] [CrossRef]
- Alekseeva, M.; Otyuskaya, D.S.; Rekhtina, M.A.; Bulavchenko, O.A.; Stonkus, O.A.; Kaichev, V.V.; Zavarukhin, S.G.; Thybaut, J.W.; Alexiadis, V.; Venderbosch, R.H. NiCuMo-SiO2 catalyst for pyrolysis oil upgrading: Model acidic treatment study. Appl. Catal. A Gen. 2019, 573, 1–12. [Google Scholar] [CrossRef]
- Smirnov, A.A.; Khromova, S.A.; Bulavchenko, O.A.; Kaichev, V.V.; Saraev, A.A.; Reshetnikov, S.I.; Bykova, M.V.; Trusov, L.I.; Yakovlev, V.A. Effect of the Ni/Cu ratio on the composition and catalytic properties of nickel-copper alloy in anisole hydrodeoxygenation. Kinet. Catal. 2014, 55, 69–78. [Google Scholar] [CrossRef]
- Callister, D.W.; Rethwisch, D.G. Materials Science and Engineering: An Introduction; John wiley & sons: New York, NY, USA, 2007; Volume 7. [Google Scholar]
- Youn, M.H.; Seo, J.G.; Kim, P.; Song, I.K. Role and effect of molybdenum on the performance of Ni-Mo/γ-Al2O3 catalysts in the hydrogen production by auto-thermal reforming of ethanol. J. Mol. Catal. A Chem. 2007, 261, 276–281. [Google Scholar] [CrossRef]
- Bang, S.; Hong, E.; Baek, S.W.; Shin, C.H. Effect of acidity on Ni catalysts supported on P-modified Al2O3 for dry reforming of methane. Catal. Today 2018, 303, 100–105. [Google Scholar] [CrossRef]
- Ravenelle, R.M.; Copeland, J.R.; Kim, W.G.; Crittenden, J.C.; Sievers, C. Structural changes of γ-Al2O3-supported catalysts in hot liquid water. ACS Catal. 2011, 1, 552–561. [Google Scholar] [CrossRef]
- Abi Aad, J.; Courty, P.; Decottignies, D.; Michau, M.; Diehl, F.; Carrier, X.; Marceau, E. Inhibition by Inorganic Dopants of γ-Alumina Chemical Weathering under Hydrothermal Conditions: Identification of Reactive Sites and their Influence in Fischer–Tropsch Synthesis. ChemCatChem 2017, 9, 2106–2117. [Google Scholar] [CrossRef] [Green Version]
- Wen, G.; Xu, Y.; Ma, H.; Xu, Z.; Tian, Z. Production of hydrogen by aqueous-phase reforming of glycerol. Int. J. Hydrogen Energy 2008, 33, 6657–6666. [Google Scholar] [CrossRef]
- Freitas, I.C.; Manfro, R.L.; Souza, M.M. Hydrogenolysis of glycerol to propylene glycol in continuous system without hydrogen addition over Cu-Ni catalysts. Appl. Catal. B Environ. 2018, 220, 31–41. [Google Scholar] [CrossRef]
- Stanislaus, A.; Absi-Halabi, M.; Al-Doloma, K. Effect of phosphorus on the acidity of γ-alumina and on the thermal stability of γ-alumina supported nickel—molybdenum hydrotreating catalysts. Appl. Catal. 1988, 39, 239–253. [Google Scholar] [CrossRef]
- Manfro, R.L.; Pires, T.P.; Ribeiro, N.F.; Souza, M.M. Aqueous-phase reforming of glycerol using Ni–Cu catalysts prepared from hydrotalcite-like precursors. Catal. Sci. Technol. 2013, 3, 1278–1287. [Google Scholar] [CrossRef]
- Bang, Y.; Han, S.J.; Yoo, J.; Choi, J.H.; Lee, J.K.; Song, J.H.; Lee, J.; Song, I.K. Hydrogen production by steam reforming of simulated liquefied natural gas (LNG) over nickel catalyst supported on mesoporous phosphorus-modified alumina xerogel. Appl. Catal. B Environ. 2014, 148, 269–280. [Google Scholar] [CrossRef]
- Salagre, P.; Fierro, J.L.; Medina, F.; Sueiras, J.E. Characterization of nickel species on several γ-alumina supported nickel samples. J. Mol. Catal. A Chem. 1996, 106, 125–134. [Google Scholar] [CrossRef]
- Thyssen, V.V.; Maia, T.A.; Assaf, E.M. Cu and Ni Catalysts supported on γ-Al2O3 and SiO2 assessed in glycerol steam reforming reaction. J. Braz. Chem. Soc. 2015, 26, 22–31. [Google Scholar]
- Yang, R.; Li, X.; Wu, J.; Zhang, X.; Zhang, Z.; Cheng, Y.; Guo, J. Hydrotreating of crude 2-ethylhexanol over Ni/Al2O3 catalysts: Surface Ni species-catalytic activity correlation. Appl. Catal. A Gen. 2009, 368, 105–112. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, N.D.; Baek, J.; Kim, W.; Lee, H.J.; Yi, J. Effect of N2O-mediated calcination on nickel species and the catalytic activity of nickel catalysts supported on γ-Al2O3 in the steam reforming of glycerol. Int. J. Hydrogen Energy 2011, 36, 3844–3852. [Google Scholar] [CrossRef]
- Bang, Y.; Han, S.J.; Seo, J.G.; Youn, M.H.; Song, J.H.; Song, I.K. Hydrogen production by steam reforming of liquefied natural gas (LNG) over ordered mesoporous nickel–alumina catalyst. Int. J. Hydrogen Energy 2012, 37, 17967–17977. [Google Scholar] [CrossRef]
- Dussault, L.; Dupin, J.C.; Dumitriu, E.; Auroux, A.; Guimon, C. Microcalorimetry, TPR and XPS studies of acid–base properties of NiCuMgAl mixed oxides using LDHs as precursors. Thermochim. Acta 2005, 434, 93–99. [Google Scholar] [CrossRef]
- Carrero, A.; Calles, J.A.; Vizcaíno, A.J. Hydrogen production by ethanol steam reforming over Cu-Ni/SBA-15 supported catalysts prepared by direct synthesis and impregnation. J. Mol. Catal. A Chem. 2007, 327, 82–94. [Google Scholar] [CrossRef]
- Valencia, D.; Klimova, T. Effect of the support composition on the characteristics of NiMo and CoMo/(Zr) SBA-15 catalysts and their performance in deep hydrodesulfurization. Catal. Today 2011, 166, 91–101. [Google Scholar] [CrossRef]
- Cordero, R.L.; Agudo, A.L. Effect of water extraction on the surface properties of Mo/Al2O3 and NiMo/Al2O3 hydrotreating catalysts. J. Mol. Catal. A Chem. 2000, 202, 23–35. [Google Scholar] [CrossRef]
- Yurdakul, M.; Ayas, N.; Bizkarra, K.; EI Doukkali, M.; Cambra, J.F. Preparation of Ni-based catalysts to produce hydrogen from glycerol by steam reforming process. Int. J. Hydrogen Energy 2016, 41, 8084–8091. [Google Scholar] [CrossRef]
- Hu, G.; Deng, X.; Pneg, Z.; Du, K. Comparison of AlPO4-and Co3(PO4)2-coated LiNi0.8Co0.2O2 cathode materials for Li-ion battery. Electrochim. Acta 2008, 53, 2567–2573. [Google Scholar] [CrossRef]
- Yuan, C.; Jiang, Y.; Wang, Z.; Xie, X.; Yang, Z.; Yousaf, A.B.; Xu, A. Cobalt phosphate nanoparticles decorated with nitrogen-doped carbon layers as highly active and stable electrocatalysts for the oxygen evolution reaction. J. Mater. Chem. A 2016, 4, 8155–8160. [Google Scholar] [CrossRef]
- Batista, J.; Pintar, A.; Mandrino, D.; Jenko, M.; Martin, V. XPS and TPR examinations of γ-alumina-supported Pd-Cu catalysts. Appl. Catal. A Gen. 2001, 206, 113–124. [Google Scholar] [CrossRef]
- Richter, M.; Fait, M.J.; Eckelt, R.; Schneider, M.; Radnik, J.; Heidemann, D.; Fricke, R. Gas-phase carbonylation of methanol to dimethyl carbonate on chloride-free Cu-precipitated zeolite Y at normal pressure. J. Catal. 2007, 245, 11–24. [Google Scholar] [CrossRef]
- Strohmeier, B.R.; Levden, D.E.; Field, R.S.; Hercules, D.M. Surface spectroscopic characterization of CuAl2O3 catalysts. J. Catal. 1985, 94, 514–530. [Google Scholar] [CrossRef]
- Wöllner, A.; Lange, F.; Schmelz, H.; Knözinger, H. Characterization of mixed copper-manganese oxides supported on titania catalysts for selective oxidation of ammonia. Appl. Catal. A Gen. 1993, 94, 181–203. [Google Scholar]
- Galtayries, A.; Wisniewski, S.; Grimblot, J. Formation of thin oxide and sulphide films on polycrystalline molybdenum foils: Characterization by XPS and surface potential variations. J. Electron Spectrosc. Relat. Phenom. 1997, 87, 31–44. [Google Scholar] [CrossRef]
- Óvári, L.; Kiss, J.; Farkas, A.P.; Solymosi, F. Surface and subsurface oxidation of Mo2C/Mo (100): Low-energy ion-scattering, auger electron, angle-resolved X-ray photoelectron, and mass spectroscopy studies. J. Phys. Chem. B 2005, 109, 4638–4645. [Google Scholar] [CrossRef] [Green Version]
- Puron, H.; Pinilla, J.L.; Saraev, A.A.; Kaichev, V.V.; Marcos, M. Hydroprocessing of Maya vacuum residue using a NiMo catalyst supported on Cr-doped alumina. Fuel 2020, 263, 116717. [Google Scholar] [CrossRef]
- Gutierrez, A.; Kaila, R.K.; Honkela, M.L.; Slioor, R.; Krause, A.O. Hydrodeoxygenation of guaiacol on noble metal catalysts. Catal. Today 2009, 147, 239–246. [Google Scholar] [CrossRef]
- He, Y.; Bie, Y.; Lehtonen, J.; Liu, R.; Cai, J. Hydrodeoxygenation of guaiacol as a model compound of lignin-derived pyrolysis bio-oil over zirconia-supported Rh catalyst: Process optimization and reaction kinetics. Fuel 2019, 239, 1015–1027. [Google Scholar] [CrossRef]
- Damyanova, S.; Pawelec, B.; Arishtirova, K.; Fierro, J.L. Biogas reforming over bimetallic PdNi catalysts supported on phosphorus-modified alumina. Int. J. Hydrogen Energy 2011, 36, 10635–10647. [Google Scholar] [CrossRef]
- Denmark, S.E.; Beutner, G.L. Lewis base catalysis in organic synthesis. Angew. Chem. Int. Ed. 2008, 47, 1560–1638. [Google Scholar] [CrossRef]
- Spojakina, A.; Damyanova, S.; Petrov, L.; Vit, Z. Effect of phosphorus on the surface state of alumina-supported nickel molybdenum catalysts for hydrodesulphurization. Appl. Catal. 1989, 56, 163–176. [Google Scholar] [CrossRef]
- Liu, F.; Okolie, C.; Ravenelle, R.M.; Crittenden, J.C.; Sievers, C.; Bruijnincx, P.C.; Weckhuysen, B.M. Silica deposition as an approach for improving the hydrothermal stability of an alumina support during glycerol aqueous phase reforming. Appl. Catal. A Gen. 2018, 551, 13–22. [Google Scholar] [CrossRef]
- Ardiyanti, A.; Khromova, S.A.; Venderbosch, R.H.; Yakovlev, V.A.; Heeres, H.J. Catalytic hydrotreatment of fast-pyrolysis oil using non-sulfided bimetallic Ni-Cu catalysts on a δ-Al2O3 support. Appl. Catal. B Environ. 2012, 117, 105–117. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, X.; Wang, Y. Catalytic and DRIFTS Studies of Pt-Based Bimetallic Alloy Catalysts in Aqueous-Phase Reforming of Glycerol. Ind. Eng. Chem. Res. 2019, 58, 2749–2758. [Google Scholar] [CrossRef]
- Dietrich, P.J.; Wu, T.; Sumer, A.; Dumesic, J.A.; Jellinek, J.; Delgass, W.N.; Ribeiro, F.H.; Miller, J.T. Aqueous phase glycerol reforming with Pt and PtMo bimetallic nanoparticle catalysts: The role of the Mo promoter. Top. Catal. 2013, 56, 1814–1828. [Google Scholar] [CrossRef]










{kind=link}
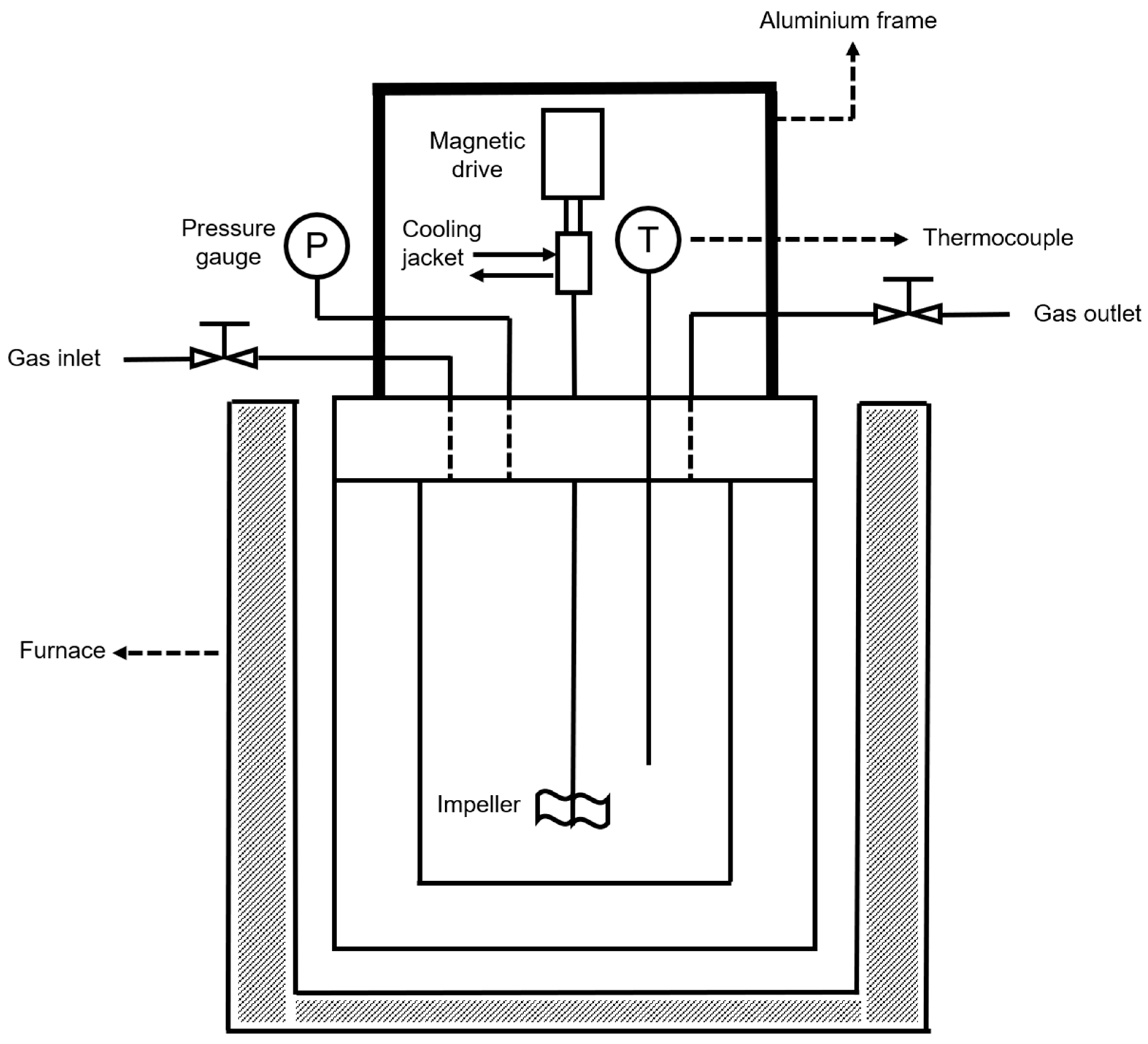{kind=link}
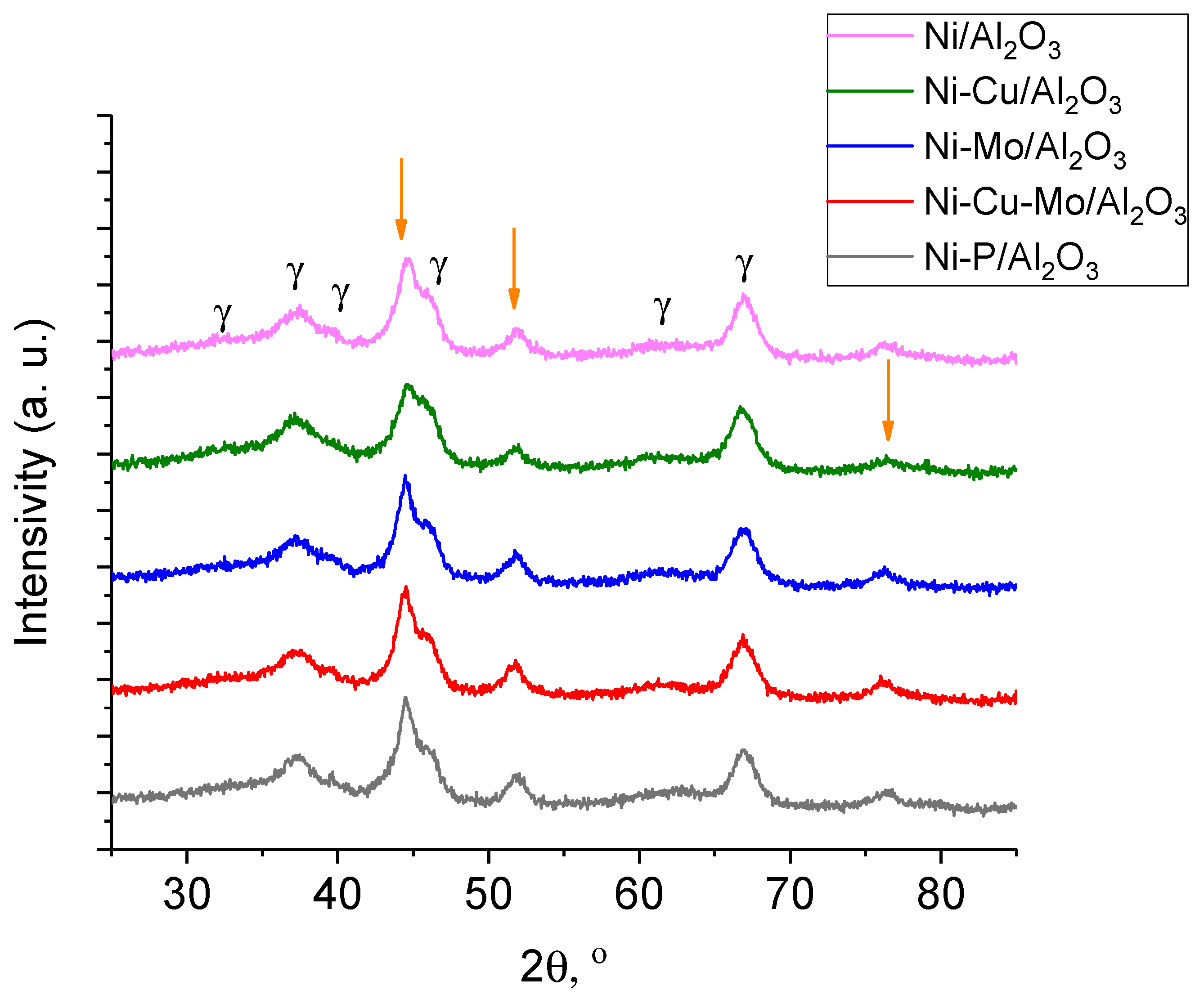{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET, m2/g | Pore Volume, cm3 g−1 | Average Pore Diameter, nm |
|---|---|---|---|
| Ni/γ-Al2O3 | 150 | 0.36 | 9.7 |
| Ni-Cu/γ-Al2O3 | 160 | 0.37 | 9.4 |
| Ni-Mo/γ-Al2O3 | 162 | 0.38 | 9.3 |
| Ni-Cu-Mo/γ-Al2O3 | 150 | 0.35 | 9.3 |
| Ni-P/γ-Al2O3 | 111 | 0.28 | 10.2 |
| Sample | Crystallite Size of Ni for Fresh Catalysts, nm | Crystallite Size of Ni for Spent Catalysts, nm | Ni Lattice Parameter for Fresh Catalysts, Å |
|---|---|---|---|
| Ni/γ-Al2O3 | 5.7 | 40.6 | 3.523 |
| Ni-Cu/γ-Al2O3 | 5.4 | 36.5 | 3.531 |
| Ni-Mo/γ-Al2O3 | 6.8 | 20.9 | 3.543 |
| Ni-Cu-Mo/γ-Al2O3 | 6.4 | 9.4 | 3.542 |
| Ni-P/γ-Al2O3 | 6.9 | 35.9 | 3.529 |
| Sample * | Al2p | Ni2p3/2 | Cu2p3/2 | Mo3d5/2 | P2p | Ni/Al | Cu/Al | Mo/Al | P/Al | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Al2O3 | Ni0 | Ni2+ | Mo0 | Mon+ | |||||||
| Ni/Al2O3 | 74.5 | 852.7 | 857.0 | – | – | – | – | 0.025 a 0.498 b | – | – | – |
| Ni-P/Al2O3 | 74.5 | 852.7 | 857.0 | – | – | – | 134.5 | 0.038 a 0.574 b | – | – | 0.034 a 0.035 b |
| Ni-Cu/Al2O3 | 74.5 | 852.7 | 857.0 | 932.3 | – | – | – | 0.023 a 0.474 b | 0.0056 a 0.0123 b | – | – |
| Ni-Mo/ Al2O3 | 74.5 | 852.8 | 856.9 | – | 227.7 | 232.2 | – | 0.025 a 0.461 b | – | 0.0039 a 0.0343 b | – |
| Ni-Cu-Mo/Al2O3 | 74.5 | 851.8 | 856.2 | 931.4 | 227.3 | 232.5 | – | 0.029 a 0.476 b | 0.0011 a 0.0166 b | 0.0066 a 0.0361 b | – |
| Product Selectivity | ||||
|---|---|---|---|---|
| Ni/Al2O3 | Ni-P/Al2O3 | Ni-Cu/Al2O3 | Ni-Mo/Al2O3 | |
| Cyclohexanol | 45% | 20% | 17% | 11% |
| Cyclohexanone | 25% | 25% | 15% | 14% |
| Phenol | 5% | 34% | 39% | 37% |
| 1-Methyl-1,2-cyclohexanediol | 15% | 8% | 19% | 10% |
| Net H2 Production (mmol H2) | H2 consumed in Guaiacol Hydrogenation (mmol H2) | CH4 Production (mmol CH4) | H2 Consumed in CH4 Production (mmol H2) | Total H2 Production (mmol H2) | H2 Selectivity to CH4 | |
|---|---|---|---|---|---|---|
| Ni/Al2O3 | 7.5 | 34.6 | 6.9 | 27.6 | 69.7 | 39.6% |
| Ni-P/Al2O3 | 7.1 | 12.7 | 7.4 | 29.6 | 49.4 | 60.0% |
| Ni-Cu/Al2O3 | 9.1 | 10.0 | 2.8 | 11.2 | 30.3 | 36.5% |
| Ni-Mo/Al2O3 | 6.0 | 4.1 | 5.6 | 22.4 | 32.5 | 68.9% |
| Ni/Al2O3 | Ni-P/Al2O3 | Ni-Cu/Al2O3 | Ni-Mo/Al2O3 | Ni-Cu-Mo/Al2O3 | |
|---|---|---|---|---|---|
| Coke yield, wt.% | 7.7 | 6.8 | 7.6 | 7.1 | 7.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Kukushkin, R.G.; Yeletsky, P.M.; Saraev, A.A.; Bulavchenko, O.A.; Millan, M. Coupling Hydrogenation of Guaiacol with In Situ Hydrogen Production by Glycerol Aqueous Reforming over Ni/Al2O3 and Ni-X/Al2O3 (X = Cu, Mo, P) Catalysts. Nanomaterials 2020, 10, 1420. https://0-doi-org.brum.beds.ac.uk/10.3390/nano10071420
Chen Z, Kukushkin RG, Yeletsky PM, Saraev AA, Bulavchenko OA, Millan M. Coupling Hydrogenation of Guaiacol with In Situ Hydrogen Production by Glycerol Aqueous Reforming over Ni/Al2O3 and Ni-X/Al2O3 (X = Cu, Mo, P) Catalysts. Nanomaterials. 2020; 10(7):1420. https://0-doi-org.brum.beds.ac.uk/10.3390/nano10071420
Chicago/Turabian StyleChen, Ziyin, Roman G. Kukushkin, Petr M. Yeletsky, Andrey A. Saraev, Olga A. Bulavchenko, and Marcos Millan. 2020. "Coupling Hydrogenation of Guaiacol with In Situ Hydrogen Production by Glycerol Aqueous Reforming over Ni/Al2O3 and Ni-X/Al2O3 (X = Cu, Mo, P) Catalysts" Nanomaterials 10, no. 7: 1420. https://0-doi-org.brum.beds.ac.uk/10.3390/nano10071420