A Theoretical Study of the Occupied and Unoccupied Electronic Structure of High- and Intermediate-Spin Transition Metal Phthalocyaninato (Pc) Complexes: VPc, CrPc, MnPc, and FePc

 , , and
, , and
Abstract
:
1. Introduction
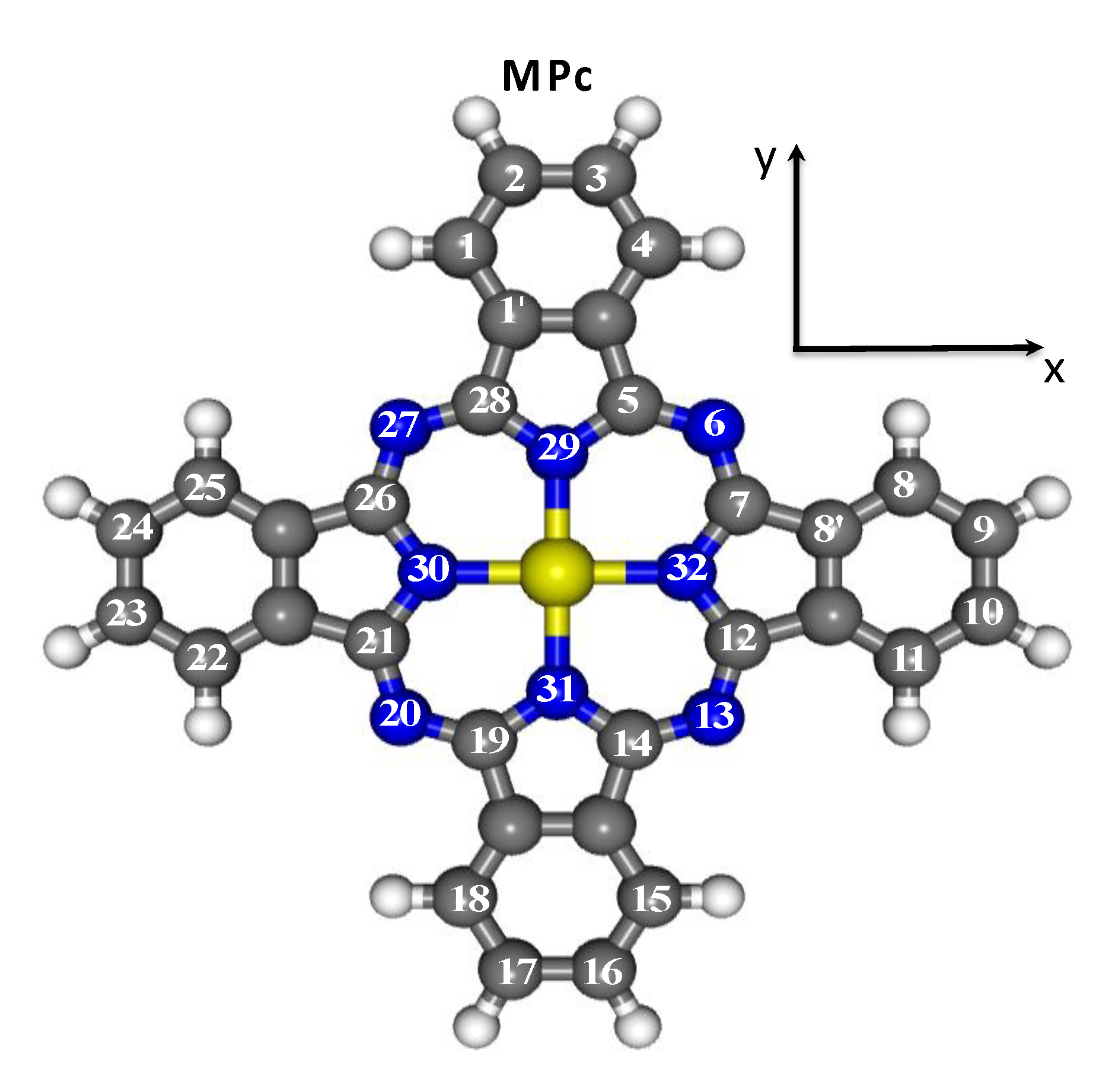
2. Computational Details
3. Results and Discussion
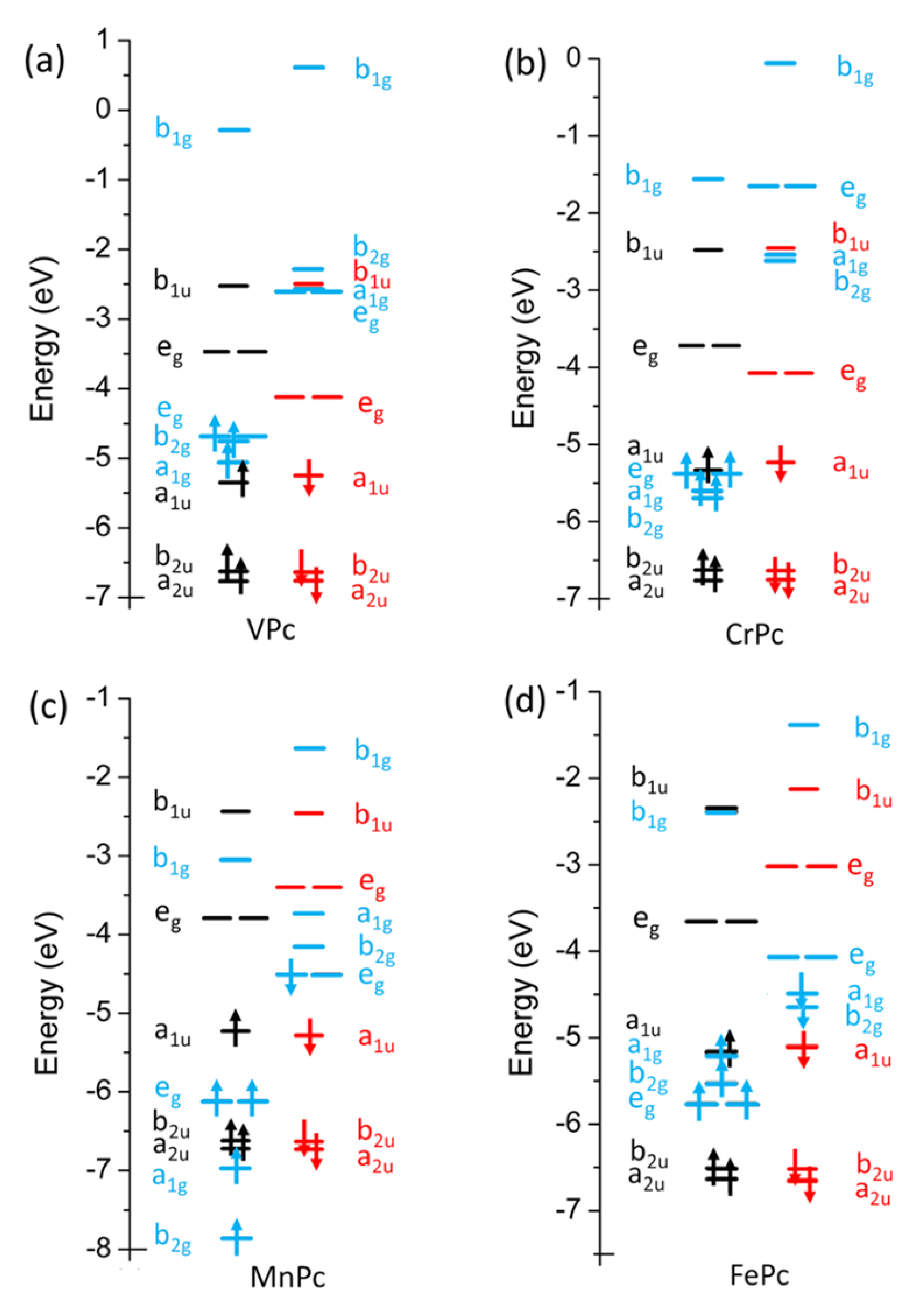
3.1. MPc Occupied Electronic Structure
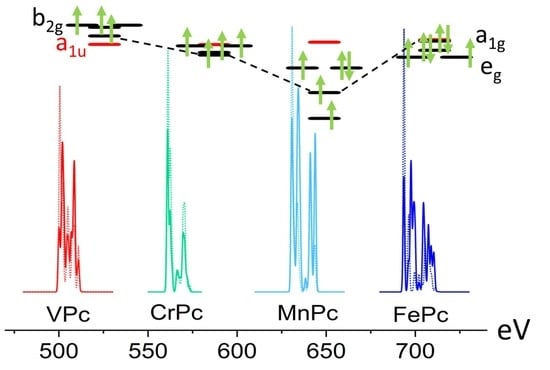
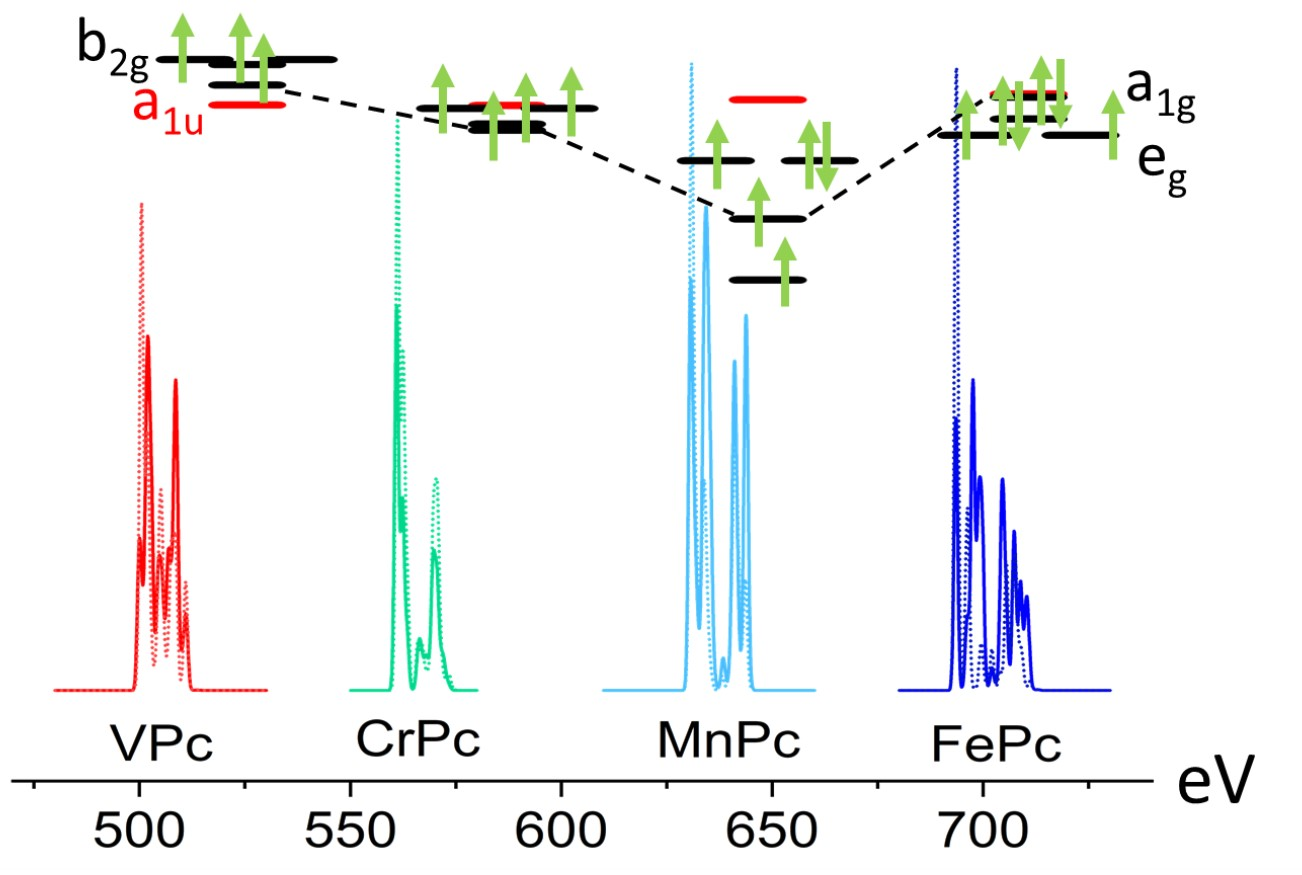
3.2. MPc Unoccupied Electronic Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghosh, A. Letters to a Young Chemist; John Wiley & Sons: Hoboken, NJ, USA, 2011; p. 34. [Google Scholar]
- Functional Phthalocyanine Molecular Materials; Springer: Berlin/Heidelberg, Germany, 2010; Volume 135.
- Wöhrle, D.; Schnurpfeil, G.; Makarov, S.G.; Kazarin, A.; Suvorova, O.N. Practical Applications of Phthalocyanines—From Dyes and Pigments to Materials for Optical, Electronic and Photo-electronic Devices. Macroheterocycles 2012, 5, 191–202. [Google Scholar] [CrossRef]
- Angione, M.D.; Pilolli, R.; Cotrone, S.; Magliulo, M.; Mallardi, A.; Palazzo, G.; Sabbatini, L.; Fine, D.; Dodabalapur, A.; Cioffi, N.; et al. Carbon based materials for electronic bio-sensing. Mater. Today 2011, 14, 424–433. [Google Scholar] [CrossRef]
- Jang, S.H.; Jen, A.K.Y. Structured Organic Non-Linear Optics. Compr. Nanosci. Technol. 2011, 1, 143–187. [Google Scholar]
- Nguyen, T. Polymer-based nanocomposites for organic optoelectronic devices. Surf. Coat. Technol. 2011, 206, 742–752. [Google Scholar] [CrossRef]
- Hains, A.W.; Liang, Z.; Woodhouse, M.A.; Gregg, B.A. Molecular Semiconductors in Organic Photovoltaic Cells. Chem. Rev. 2010, 110, 6689–6735. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Xue, J. Recent progress in organic photovoltaics: Device architecture and optical design. Energy Environ. Sci. 2014, 7, 2123–2144. [Google Scholar] [CrossRef]
- Sorokin, A.B. Phthalocyanine Metal Complexes in Catalysis. Chem. Rev. 2013, 113, 8152–8191. [Google Scholar] [CrossRef]
- Tolman, W.B.; Solomon, E.I. Preface: Forum on Dioxygen Activation and Reduction. Inorg. Chem. 2010, 49, 3555–3556. [Google Scholar] [CrossRef]
- Scherson, D.A.; Palencsár, A.; Tolmachev, Y.; Stefan, I. Transition Metal Macrocycles as Electrocatalysts for Dioxygen Reduction. In Electrochemical Surface Modification: Thin Films, Functionalization and Characterization; Alkire, R.C., Kolb, D.M., Lipkowski, J., Ross, P.N., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 191–288. [Google Scholar]
- Sedona, F.; Di Marino, M.; Forrer, D.; Vittadini, A.; Casarin, M.; Cossaro, A.; Floreano, L.; Verdini, A.; Sambi, M. Tuning the Catalytic Activity of Ag(110)-supported Fe Phthalocyanine in the Oxygen Reduction Reaction. Nat. Mater. 2012, 11, 970–977. [Google Scholar] [CrossRef]
- Bianconi, A. XANES Spectroscopy. In X-ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS and XANES; Koningsberger, D.C., Prins, R., Eds.; John Wiley & Sons: New York, NY, USA, 1988; pp. 573–662. [Google Scholar]
- Stöhr, J. NEXAFS Spectroscopy; Springer: Berlin, Germany, 1996. [Google Scholar]
- Zhang, H.H.; Hedman, B.; Hodgson, K.O. X-ray Absorption Spectroscopy and EXAFS Analysis. The Multiple-Scattering Method and Applications in Inorganic and Bioinorganic Chemistry. In Inorganic Electronic Structure and Spectroscopy; Solomon, E.I., Lever, A.B.P., Eds.; John Wiley & Sons: New York, NY, USA, 1999; Volume 1, pp. 513–554. [Google Scholar]
- Solomon, E.I.; Bell, C.B., III. Inorganic and Bioinorganic Spectroscopy. In Physical Inorganic Chemistry, Principle, Methods and Models; Bakac, A., Ed.; John Wiley & Sons: New York, NY, USA, 2010; pp. 1–37. [Google Scholar]
- Douglas, B.E.; Hollingsworth, C.A. Symmetry in Bonding and Spectra, an Introduction; Academic Press: Orlando, FL, USA, 1985. [Google Scholar]
- Neese, F.; Hedman, B.; Hodgson, K.O.; Solomon, E.I. Relationship between the Dipole Strength of Ligand Pre-Edge Transitions and Metal−Ligand Covalency. Inorg. Chem. 1999, 38, 4854–4860. [Google Scholar] [CrossRef]
- Glaser, T.; Hedman, B.; Hodgson, L.O.; Solomon, E.I. Ligand K-Edge X-ray Absorption Spectroscopy: A Direct Probe of Ligand−Metal Covalency. Acc. Chem. Res. 2000, 33, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Hedman, B.; Hodgson, K.O.; Dey, A.; Szilagyi, R.K. Ligand K-edge x-ray absorption spectroscopy: Covalency of ligand-metal bonds. Coord. Chem. Rev. 2005, 249, 97–129. [Google Scholar] [CrossRef]
- Baker, M.-L.; Mara, M.W.; Yan, J.J.; Hodgson, K.O.; Hedman, B.; Solomon, E.I. K- And L-edge X-ray Absorption Spectroscopy (XAS) and Resonant Inelastic X-ray Scattering (RIXS) Determination of Differential Orbital Covalency (DOC) of Transition Metal Sites. Coord. Chem. Rev. 2017, 345, 182–208. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, C.K. Absorption Spectra and Chemical Bonding in Complexes; Pergamon Press: Oxford, UK, 1962; p. 77. [Google Scholar]
- de Groot, F. Multiplet effects in X-ray spectroscopy. Coord. Chem. Rev. 2005, 249, 31–63. [Google Scholar] [CrossRef]
- de Groot, F.; Kotani, A. Core Level Spectroscopy of Solids; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Roemelt, M.; Neese, F. Excited States of Large Open-Shell Molecules: An Efficient, General, and Spin-Adapted Approach Based on a Restricted Open-Shell Ground State Wave function. J. Phys. Chem. A 2013, 117, 3069–3083. [Google Scholar] [CrossRef]
- Bagus, P.S.; Freund, H.; Kuhlenbeck, H.; Ilton, E.S. A new analysis of X-ray adsorption branching ratios: Use of Russell−Saunders coupling. Chem. Phys. Lett. 2008, 455, 331–334. [Google Scholar] [CrossRef]
- Ikeno, H.; Mizoguchi, T.; Tanaka, I. Ab initio charge transfer multiplet calculations on the L2,3 XANES and ELNES of 3d transition metal oxides. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 83, 155107. [Google Scholar] [CrossRef] [Green Version]
- Josefsson, I.; Kunnus, K.; Schreck, S.; Föhlisch, A.; de Groot, F.; Wernet, P.; Odelius, M. Ab Initio calculations of x-ray spectra: Atomic multiplet and molecular orbital effects in a multiconfigurational SCF approach to the L-edge spectra of transition metal complexes. J. Phys. Chem. Lett. 2012, 3, 3565–3570. [Google Scholar] [CrossRef] [Green Version]
- Maganas, D.; Roemelt, M.; Hävecker, M.; Trunschke, A.; Knop-Gericke, A.; Schlögl, R.; Neese, F. First principles calculations of the structure and V L-edge X-ray absorption spectra of V2O5 using local pair natural orbital coupled cluster theory and spin−orbit coupled configuration interaction approaches. Phys. Chem. Chem. Phys. 2013, 15, 7260–7276. [Google Scholar] [CrossRef] [Green Version]
- Roemelt, M.; Maganas, D.; DeBeer, S.; Neese, F. A combined DFT and restricted open-shell configuration interaction method including spin-orbit coupling: Application to transition metal L-edge X-ray absorption spectroscopy. J. Chem. Phys. 2013, 138, 204101 (1:22). [Google Scholar] [CrossRef]
- Maganas, D.; DeBeer, S.; Neese, F. Restricted open-shell configuration interaction cluster calculations of the L-edge x-ray absorption study of TiO2 and CaF2 solids. Inorg. Chem. 2014, 53, 6374–6385. [Google Scholar] [CrossRef] [PubMed]
- Maganas, D.; Roemelt, M.; Weyhermüller, T.; Blume, R.; Hävecker, M.; Knop-Gericke, A.; DeBeer, S.; Schlögl, R.; Neese, F. L-edge X-ray absorption study of mononuclear vanadium complexes and spectral predictions using a restricted open shell configuration interaction ansatz. Phys. Chem. Chem. Phys. 2014, 16, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshino, M.; Kurata, H.; Isoda, S.; Kobayashi, T. Branching ratio and L2 + L3 intensities of 3d-transition metals in phthalocyanines and the amine complexes. Micron 2000, 31, 373–380. [Google Scholar] [CrossRef]
- Kroll, T.; Kraus, R.; Schönfelder, R.; Aristov, V.Y.; Molodtsova, O.V.; Hoffmann, P.; Knupfer, M. Transition metal phthalocyanines: Insight into the electronic structure from soft x-ray spectroscopy. J. Chem. Phys. 2012, 137, 054306. [Google Scholar] [CrossRef]
- Eguchi, K.; Nakagawa, T.; Takagi, Y.; Yokoyama, T. Direct Synthesis of Vanadium Phthalocyanine and Its Electronic and Magnetic States in Monolayers and Multilayers on Ag(111). J. Phys. Chem. C 2015, 119, 9805–9815. [Google Scholar] [CrossRef]
- Carlotto, S.; Sambi, M.; Rancan, M.; Casarin, M. Theoretical Investigation of the Electronic Properties of Three Vanadium Phthalocyaninato (Pc) Based Complexes: PcV, PcVO, and PcVI. Inorg. Chem. 2018, 57, 1859–1869. [Google Scholar] [CrossRef]
- Casarin, M.; Carlotto, S. Pigments of Life”, Molecules Well Suited to Investigate Metal–Ligand Symmetry-Restricted Covalency. Eur. J. Inorg. Chem. 2018, 3145–3155. [Google Scholar] [CrossRef]
- Carlotto, S.; Sambi, M.; Sedona, F.; Vittadini, A.; Bartolomé, J.; Bartolomé, F.; Casarin, M. L2,3-edges absorption spectra of a 2D complex system: A theoretical modelling. Phys. Chem. Chem. Phys. 2016, 18, 28110–28116. [Google Scholar] [CrossRef]
- Nardi, M.V.; Detto, F.; Aversa, L.; Verucchi, R.; Salviati, G.; Iannotta, S.; Casarin, M. Electronic properties of CuPc and H2Pc: An experimental and theoretical study. Phys. Chem. Chem. Phys. 2013, 15, 12864–12881. [Google Scholar] [CrossRef]
- Mangione, G.; Sambi, M.; Nardi, M.V.; Casarin, M. A theoretical study of the L3 pre-edge XAS in Cu(II) complexes. Phys. Chem. Chem. Phys. 2014, 16, 19852–19855. [Google Scholar] [CrossRef]
- Mangione, G.; Sambi, M.; Carlotto, S.; Vittadini, A.; Ligorio, G.; Timpel, M.; Pasquali, L.; Giglia, A.; Nardi, M.V.; Casarin, M. Electronic structure of CuTPP and CuTPP(F) complexes: A combined experimental and theoretical study II. Phys. Chem. Chem. Phys. 2016, 18, 24890–24904. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wiley. The ORCA program system. Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Wang, F.; Ziegler, T. The calculation of excitation energies based on the relativistic two-component zeroth-order regular approximation and time-dependent density-functional with full use of symmetry. J. Chem. Phys. 2005, 122, 204103. [Google Scholar] [CrossRef] [PubMed]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Carlotto, S.; Floreano, L.; Cossaro, A.; Dominguez, M.; Rancan, M.; Sambi, M.; Casarin, M. The electronic properties of three popular high spin complexes [TM(acac)3, TM = Cr, Mn, and Fe] revisited: An experimental and theoretical study. Phys. Chem. Chem. Phys. 2017, 19, 24840–24854. [Google Scholar] [CrossRef]
- Casarin, M.; Finetti, P.; Vittadini, A.; Wang, F.; Ziegler, T. Spin-Orbit Relativistic Time-Dependent Density Functional Calculations of the Metal and Ligand Pre-Edge XAS Intensities of Organotitanium Complexes: TiCl4, Ti(η5-C5H5)Cl3, and Ti(η5-C5H5)2Cl2. J. Phys. Chem. A 2007, 111, 5270–5279. [Google Scholar] [CrossRef]
- Carlotto, S.; Finetti, P.; de Simone, M.; Coreno, M.; Casella, G.; Sambi, M.; Casarin, M. Comparative Experimental and Theoretical Study of the C and O K-Edge X-ray Absorption Spectroscopy in Three Highly Popular, Low Spin Organoiron Complexes: [Fe(CO)5], [(η5-C5H5)Fe(CO)(μ-CO)]2, and [(η5-C5H5)2Fe]. Inorg. Chem. 2019, 58, 16411–16423. [Google Scholar] [CrossRef]
- Carlotto, S.; Finetti, P.; de Simone, M.; Coreno, M.; Casella, G.; Sambi, M.; Casarin, M. Comparative Experimental and Theoretical Study of the Fe L2,3-Edges X-ray Absorption Spectroscopy in Three Highly Popular, Low Spin Organoiron Complexes: [Fe(CO)5], [(η5-C5H5)Fe(CO)(μ-CO)]2, and [(η5-C5H5)2Fe]. Inorg. Chem. 2019, 58, 5844–5857. [Google Scholar] [CrossRef]
- Wu, W.; Harrison, N.M.; Fisher, A.J. Suitability of chromium phthalocyanines to test Haldane’s conjecture: First-principles calculations. Phys. Rev. B: Condens. Matter Mater. Phys. 2013, 88, 224417. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Q. The superior catalytic CO oxidation capacity of a Cr-phthalocyanine porous sheet. Sci. Rep. 2014, 4, 4098. [Google Scholar] [CrossRef] [Green Version]
- Junkaew, A.; Meeprasert, J.; Jansang, B.; Kungwan, N.; Namuangruk, S. Mechanistic study of NO oxidation on Cr–phthalocyanine: Theoretical insight. RSC Adv. 2017, 7, 8858–8865. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Lei, Y.; Wang, G. Correlation between oxygen adsorption energy and electronic structure of transition metal macrocyclic complexes. J. Chem. Phys. 2013, 139, 204306. [Google Scholar] [CrossRef] [PubMed]
- Bala, M.; Verma, P.K.; Sharma, U.; Kumar, N.; Singh, B. Iron phthalocyanine as an efficient and versatile catalyst for N-alkylation of heterocyclic amines with alcohols: One-pot synthesis of 2-substituted benzimidazoles, benzothiazoles and benzoxazoles. Green Chem. 2013, 15, 1687–1693. [Google Scholar] [CrossRef]
- Paradine, S.M.; White, M.C. Iron-Catalyzed Intramolecular Allylic C–H Amination. J. Am. Chem. Soc. 2012, 134, 2036–2039. [Google Scholar] [CrossRef] [PubMed]
- Lamar, A.A.; Nicholas, K.M. Direct synthesis of 3-arylindoles via annulation of aryl hydroxylamines with alkynes. Tetrahedron 2009, 65, 3829–3833. [Google Scholar] [CrossRef]
- Taniguchi, T.; Hirose, D.; Ishibashi, H. Esterification via Iron-Catalyzed Activation of Triphenylphosphine with Air. ACS Catal. 2011, 1, 1469–1474. [Google Scholar] [CrossRef]
- Prateeptongkum, S.; Jovel, I.; Jackstell, R.; Vogl, N.; Weckbecker, C.; Beller, M. First iron-catalyzed synthesis of oximes from styrenes. Chem. Commun. 2009, 1990–1992. [Google Scholar] [CrossRef]
- Kudrik, E.; Makarov, S.V.; Zahl, A.; van Eldik, R. Mechanism of the Iron Phthalocyanine Catalyzed Reduction of Nitrite by Dithionite and Sulfoxylate in Aqueous Solution. Inorg. Chem. 2005, 44, 6470–6475. [Google Scholar] [CrossRef]
- Sugimori, T.; Horike, S.I.; Tsumura, S.; Hande, M.; Kasuga, K. Catalytic oxygenation of olefin with dioxygen and tetra-t-butylphthalocyanine complexes in the presence of sodium borohydride. Inorg. Chim. Acta 1998, 283, 275–278. [Google Scholar] [CrossRef]
- Leggans, E.K.; Barker, T.J.; Duncan, K.K.; Boger, D.L. Iron(III)/NaBH4-Mediated Additions to Unactivated Alkenes: Synthesis of Novel 20′-Vinblastine Analogues. Org. Lett. 2012, 14, 1428–1431. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, T.; Goto, N.; Nishibata, A.; Ishibashi, H. Iron-Catalyzed Redox Radical Cyclizations of 1,6-Dienes and Enynes. Org. Lett. 2010, 12, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Auwärter, W.; Écija, D.; Klappenberger, E.; Barth, J.V. Porphyrins at interfaces. Nat. Chem. 2015, 7, 105–120. [Google Scholar] [CrossRef] [PubMed]
- ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 28 December 2020).
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. A: At. Mol. Opt. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter Mater. Phys. 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J. Modified valence indices from the two-particle density matrix. Int. J. Quantum Chem. 1994, 51, 187–200. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J.; Formosinho, S.J.; Varandas, A.J.C. Quantum mechanical valence study of a bond-breaking−bond forming process in triatomic systems. Int. J. Quantum Chem. 1994, 52, 1153–1176. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J. Hartree-Fock difference approach to chemical valence: Three-electron indices in UHF approximation. Int. J. Quantum Chem. 1996, 57, 377–389. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J.; Mazur, G. Quantum chemical valence indices from the one-determinantal difference approach. Can. J. Chem. 1996, 74, 1121–1130. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J.; Michalak, A. Two-electron valence indices from the Kohn-Sham orbitals. Int. J. Quantum Chem. 1997, 61, 589–601. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J.; Michalak, A. Exploring bonding patterns of molecular systems using quantum mechanical bond multiplicities. Polym. J. Chem. 1998, 72, 1779–1791. [Google Scholar]
- Michalak, A.; DeKock, R.L.; Ziegler, T. Bond Multiplicity in Transition-Metal Complexes: Applications of Two-Electron Valence Indices. J. Phys. Chem. A 2008, 112, 7256–7263. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.J. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.Y.; Landis, C.R.; Neese, F. All-electron scalar relativistic basis sets for third-row transition metal atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef]
- Lebedev, V.I. Values of the nodes and weights of quadrature formulas of Gauss−Markov type for a sphere from the ninth to seventeenth order of accuracy that are invariant with respect to an octahedron group with inversion. Zhurnal Vychislitel’noi Mat. Mat. Fiz. 1975, 15, 48–54. [Google Scholar]
- Coster, D.; Kronig, R.D.L. A new type of Auger effect and its influence on the X-ray spectrum. Physica 1935, 2, 13–24. [Google Scholar] [CrossRef]
- Mangione, G.; Carlotto, S.; Sambi, M.; Ligorio, G.; Timpel, M.; Vittadini, A.; Nardi, M.V.; Casarin, M. Electronic structures of CuTPP and CuTPP(F) complexes. A combined experimental and theoretical study I. Phys. Chem. Chem. Phys. 2016, 18, 18727–18738. [Google Scholar] [CrossRef]
- Arillo-Flores, O.I.; Fadlallah, M.M.; Schuster, C.; Eckern, U.; Romero, A.H. Magnetic, electronic and vibrational properties of metal and fluorinated metal phthalocyanines. Phys. Rev. B: Condens. Matter Mater. Phys. 2013, 87, 165115. [Google Scholar] [CrossRef] [Green Version]
- Jahn, H.; Teller, E. Stability of polyatomic molecules in degenerate electronic states-I—Orbital degeneracy. Proc. R. Soc. London, Ser. A 1937, 161, 220–235. [Google Scholar]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Slater, J.C. Quantum Theory of Molecules and Solids. The Self-Consistent-Field for Molecules and Solids; McGraw-Hill: New York, NY, USA, 1974. [Google Scholar]
- Liberman, D.A. Slater transition-state band-structure calculations. Phys. Rev. B: Condens. Matter Mater. Phys. 2000, 62, 6851–6853. [Google Scholar] [CrossRef]
- Berkowitz, J. Photoelectron spectroscopy of phthalocyanine vapors. J. Chem. Phys. 1979, 70, 2819–2828. [Google Scholar] [CrossRef]
- A comprehensive collection of single-crystal structures of phthalocyanine complexes and related macrocycles is reported in M.K. Engel. In The Porphyrin Handbook; Phthalocyanines: Structural Characterization; Academic Press: New York, NY, USA, 2003; Volume 20, pp. 1–242.
- Elvidge, J.A.; Lever, B.P. Metal chelates. Part II. Phthalocyanine–chromium complexes and perpendicular conjugation. J. Chem. Soc. 1961, 1257–1265. [Google Scholar] [CrossRef]
- Lever, A.B.P. The magnetic behaviour of transition-metal phathalocyanines. J. Chem. Soc. 1965, 1821–1829. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef] [Green Version]
- Figgis, B.N.; Mason, R.; Williams, G.A. Structure of Phthalocyaninatomanganese(II) at 5.8 K Determined by Neutron Diffraction. Acta Cryst. 1980, B36, 2963–2970. [Google Scholar] [CrossRef]
- Kirner, J.F.; Dow, D.; Scheidt, W.R. Molecular Stereochemistry of Two Intermediate-Spin Complexes. Iron(II) Phthalocyanine and Manganese(II) Phthalocyanine. Inorg. Chem. 1976, 15, 1685–1690. [Google Scholar] [CrossRef]
- Barraclough, C.G.; Martin, R.L.; Mitra, S.; Sherwood, R.C. Paramagnetic Anisotropy, Electronic Structure, and Ferromagnetism in Spin S = 3/2 Manganese(II) Phthalocyanine. J. Chem. Phys. 1970, 53, 1638–1642. [Google Scholar] [CrossRef]
- Mitra, S.; Greson, A.K.; Hatfield, W.E.; Weller, R.R. Single-Crystal Magnetic Study on Ferromagnetic Manganese(II) Phthalocyaninate. Inorg. Chem. 1983, 22, 1729–1732. [Google Scholar] [CrossRef]
- Williamson, B.E.; van Cott, T.C.; Boyle, M.E.; Misener, C.G.; Stillman, M.J.; Schatz, P.N. Determination of the Ground State of Manganese Phthalocyanine in an Argon Matrix Using Magnetic Circular Dichroism and Absorption Spectroscopy. J. Am. Chem. Soc. 1992, 114, 2412. [Google Scholar] [CrossRef]
- Taguchi, Y.; Miyake, T.; Margadonna, S.; Kato, K.; Prassides, K.; Iwasa, Y. Synthesis, Structure, and Magnetic Properties of Li-Doped Manganese-Phthalocyanine, Lix[MnPc] (0 ≤ x ≤ 4). J. Am. Chem. Soc. 2006, 128, 3313. [Google Scholar] [CrossRef]
- Petraki, F.; Peisert, H.; Hoffmann, P.; Uihlein, J.; Knupfer, M.; Chassé, T. Modification of the 3d-Electronic Configuration of Manganese Phthalocyanine at the Interface to Gold. J. Phys. Chem. C 2012, 116, 5121–5127. [Google Scholar] [CrossRef]
- Kataoka, T.; Sakamoto, Y.; Yamazaki, Y.; Singh, V.R.; Fujimori, A.; Takeda, Y.; Ohkochic, T.; Fujimori, S.I.; Okane, T.; Saitoh, Y.; et al. Electronic configuration of Mn ions in the π-d molecular ferromagnet β-Mn phthalocyanine studied by soft X-ray magnetic circular dichroism. Solid State Commun. 2012, 152, 806–809. [Google Scholar] [CrossRef] [Green Version]
- Gottfrield, J.M. Surface chemistry of porphyrins and phthalocyanines. Surf. Sci. Reports 2015, 70, 259–379. [Google Scholar] [CrossRef]
- Liao, M.S.; Scheiner, S. Electronic structure and bonding in metal phthalocyanines, Metal = Fe, Co, Ni, Cu, Zn, Mg. J. Chem. Phys. 2001, 114, 9780–9791. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.S.; Watts, J.D.; Huang, M.J. DFT Study of Unligated and Ligated ManganeseII Porphyrins and Phthalocyanines. Inorg. Chem. 2005, 44, 1941–1949. [Google Scholar] [CrossRef]
- Marom, N.; Kronik, L. Density functional theory of transition metal phthalocyanines, II: Electronic structure of MnPc and FePc—symmetry and symmetry breaking. Appl. Phys. A 2009, 95, 165–172. [Google Scholar] [CrossRef]
- Shen, X.; Sun, L.; Benassi, E.; Shen, Z.; Zhao, X.; Sanvito, S.; Hou, S. Spin filter effect of manganese phthalocyanine contacted with single-walled carbon nanotube electrodes. J. Chem. Phys. 2010, 132, 054703. [Google Scholar] [CrossRef]
- Brumboiu, I.E.; Totani, R.; de Simone, M.; Coreno, M.; Grazioli, C.; Lozzi, L.; Herper, H.C.; Sanyal, B.; Eriksson, O.; Pugli, C.; et al. Elucidating the 3d Electronic Configuration in Manganese Phthalocyanine. J. Phys. Chem. A 2014, 118, 927–932. [Google Scholar] [CrossRef] [Green Version]
- Wäckerlin, C.; Donati, F.; Singha, A.; Baltic, R.; Uldry, A.C.; Delley, B.; Rusponi, S.; Dreiser, J. Strong antiferromagnetic exchange between manganese phthalocyanine and ferromagnetic europium oxide. Chem. Comm. 2015, 51, 12958–12961. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Shi, Y.; Cao, J.; Wu, R. Magnetization and magnetic anisotropy of metallophthalocyanine molecules from the first principles calculations. Appl. Phys. Lett. 2009, 94, 122502. [Google Scholar] [CrossRef]
- Grobosch, M.; Mahns, B.; Loose, C.; Friedrich, R.; Schmidt, C.; Kortus, J.; Knupfer, M. Identification of the electronic states of manganese phthalocyanine close to the Fermi level. Chem. Phys. Lett. 2011, 505, 122–125. [Google Scholar] [CrossRef]
- Grobosch, M.; Aristov, V.Y.; Molodtsova, O.V.; Schmidt, C.; Doyle, B.P.; Nannarone, S.; Knupfer, M. Engineering of the Energy Level Alignment at Organic Semiconductor Interfaces by Intramolecular Degrees of Freedom: Transition Metal Phthalocyanines. J. Phys. Chem. C 2009, 113, 13219–13222. [Google Scholar] [CrossRef]
- Dale, B.W.; Williams, R.J.P.; Johnson, C.E.; Thorp, T.L. S=1 Spin State of Divalent Iron. I. Magnetic Properties of Phthalocyanine Iron (II). J. Chem. Phys. 1968, 49, 3441–3444. [Google Scholar] [CrossRef]
- Dale, B.W.; Williams, R.J.P.; Johnson, C.E.; Thorp, T.L. S=1 Spin State of Divalent Iron. II. A Mössbauer-Effect Study of Phthalocyanine Iron (II). J. Chem. Phys. 1968, 49, 3445–3449. [Google Scholar] [CrossRef]
- Coppens, P.; Li, L.; Zhu, N.J. Electronic ground state of iron(II) phthalocyanine as determined from accurate diffraction data. J. Am. Chem. Soc. 1983, 105, 6173–6174. [Google Scholar] [CrossRef]
- Evangelisti, M.; Bartolomé, J.; de Jongh, L.J.; Filoti, G. Magnetic properties of α-iron(II) phthalocyanine. Phys. Rev. B Condens. Matter Mater. Phys. 2002, 66, 144410. [Google Scholar] [CrossRef] [Green Version]
- Filoti, G.; Kuz’min, M.D.; Bartolomé, J. Mössbauer study of the hyperfine interactions and spin dynamics in α-iron(II) phthalocyanin. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 74, 134420. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.S.; Scheiner, S. Comparative study of metal-porphyrins, -porphyrazines, and -phthalocyanines. J. Comput. Chem. 2002, 23, 1391–1403. [Google Scholar] [CrossRef]
- Mangione, G.; Pandolfo, L.; Sambi, M.; Ligorio, G.; Nardi, M.V.; Cossaro, A.; Floreano, L.; Casarin, M. Ligand-Field Strength and Symmetry-Restricted Covalency in Cu II Complexes—A Near-Edge X-ray Absorption Fine Structure Spectroscopy and Time-Dependent DFT Study. Eur. J. Inorg. Chem. 2015, 2709–2713. [Google Scholar]
- Nardi, M.V.; Verucchi, R.; Pasquali, L.; Giglia, A.; Fronzoni, G.; Sambi, M.; Mangione, G.; Casarin, M. XAS of tetrakis(phenyl)- and tetrakis (pentafluorophenyl)-porphyrin: An experimental and theoretical study. Phys. Chem. Chem. Phys. 2015, 17, 2001–2011. [Google Scholar] [CrossRef]
- Carlotto, S.; Sambi, M.; Vittadini, A.; Casarin, M. Theoretical modelling of the L2,3-edge X-ray absorption spectra of Mn(acac)2 and Co(acac)2 complexes. Phys. Chem. Chem. Phys. 2016, 18, 2242–2249. [Google Scholar] [CrossRef]
- Carlotto, S.; Casella, G.; Floreano, L.; Verdini, A.; Ribeiro, A.P.C.; Martins, L.M.D.R.S.; Casarin, M. Spin state, electronic structure and bonding on C-scorpionate [Fe(II)Cl2(tpm)] catalyst: An experimental and computational study. Catal. Tod. 2020, 358, 403–411. [Google Scholar] [CrossRef]
- Cojocariu, I.; Carlotto, S.; Sturmeit, H.M.; Zamborlini, G.; Cinchetti, M.; Cossaro, A.; Verdini, A.; Floreano, L.; Jugovac, M.; Puschnig, P.; et al. Ferrous to ferric transition in Fe-phthalocyanine driven by NO2 exposure. Chem.-Eur. J. in press. [CrossRef]
- Ballhausen, C.J. Introduction to Ligand Field Theory; McGraw-Hill Book Company, Inc.: New York, NY, USA, 1962; p. 69. [Google Scholar]
- Sugano, S.; Tanabe, Y.; Kamimura, H. Multiplets of Transition-Metal Ions in Crystals; Academic Press: New York, NY, USA, 1970. [Google Scholar]
- Carlotto, S.; Sambi, M.; Vittadini, A.; Casarin, M. Mn(acac)2 and Mn(acac)3 complexes, a theoretical modelling of their L2,3-edges X-ray absorption spectra. Polyhedron 2017, 135, 216–223. [Google Scholar] [CrossRef]
- Simonov, K.A.; Vinogradov, A.S.; Brzhezinskaya, M.M.; Preobrajenski, A.B.; Generalov, A.V.; Yu Klyushin, A. Features of metal atom 2p excitations and electronic structure of 3d-metal phthalocyanines studied by X-ray absorption and resonant photoemission. Appl. Surf. Sci. 2013, 267, 132–135. [Google Scholar] [CrossRef]
- Bartolomé, J.; Bartolomé, F.; Brookes, N.B.; Sedona, F.; Basagni, A.; Forrer, D.; Sambi, M. Reversible Fe Magnetic Moment Switching in Catalytic Oxygen Reduction Reaction of Fe-Phthalocyanine Adsorbed on Ag(110). J. Phys. Chem. C 2015, 119, 12488–12495. [Google Scholar] [CrossRef]
- Betti, M.G.; Gargiani, P.; Frisenda, R.; Biagi, R.; Cossaro, A.; Verdini, A.; Floreano, L.; Mariani, C. Localized and Dispersive Electronic States at Ordered FePc and CoPc Chains on Au(110). J. Phys. Chem. C 2010, 114, 21638–21644. [Google Scholar] [CrossRef]
- Stepanow, S.; Lodi Rizzini, A.; Krull, C.; Kavich, J.; Cezar, J.C.; Yakhou-Harris, F.; Sheverdyaeva, P.M.; Moras, P.; Carbone, C.; Ceballos, G.; et al. Spin Tuning of Electron-Doped Metal–Phthalocyanine Layers. J. Am. Chem. Soc. 2014, 136, 5451–5459. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
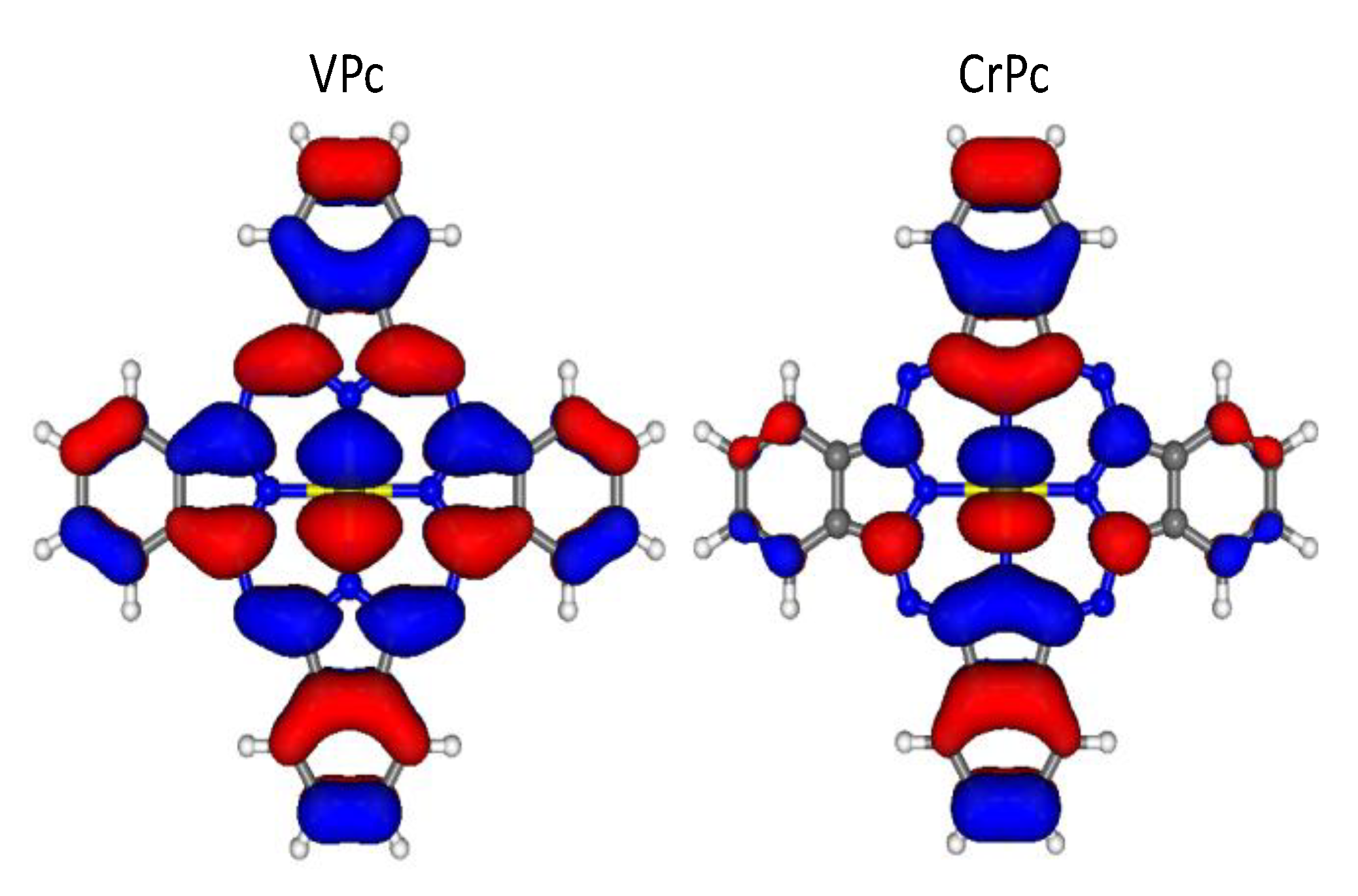{kind=link}
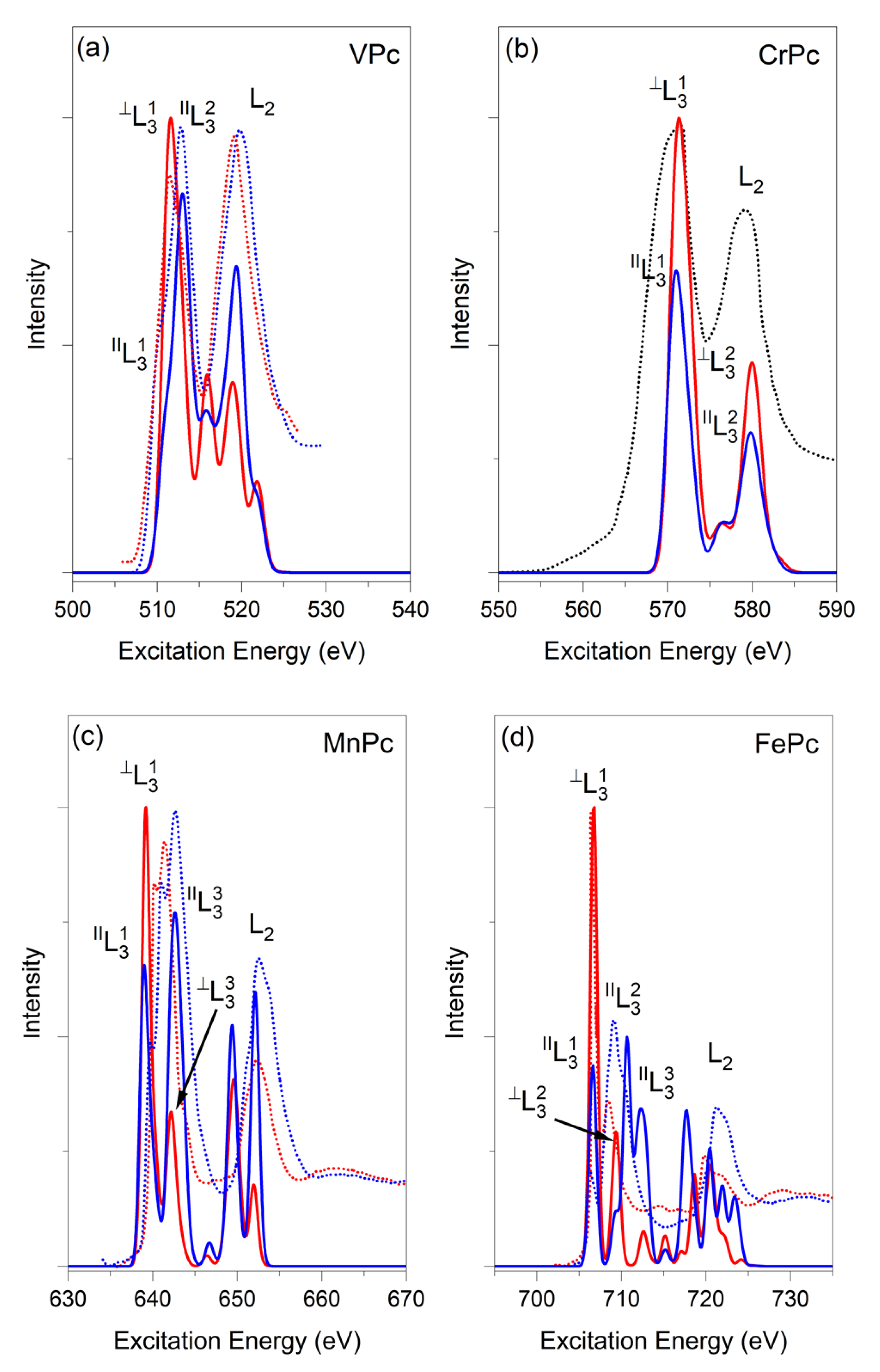{kind=link}
| S | VPc | CrPc | MnPc | FePc |
|---|---|---|---|---|
| 0 | NC a | 31.9 (1A1g) b | ||
| 1/2 | 16.6 (2B1g) | 14.9 (2B2u) | ||
| 1 | 25.6b (3Eu) | 0 (3A2g)90 | ||
| 3/2 | 0 (4Eg) | 0 (4Eg) | ||
| 2 | 0 (5B1g) | NCa | ||
| 5/2 | 16.5 (6Eg) |
| M–NPy | NPy–C | Nm–C | M–NPy–C | NPy–C–Nm | |
|---|---|---|---|---|---|
| VPc * | 1.996 | 1.392 | 1.333 | 125.4 | 127.4 |
| CrPc * | 1.982 | 1.387 | 1.330 | 125.6 | 127.5 |
| MnPc a | 1.952 (1.938) | 1.396 (1.392) | 1.324 (1.315) | 126.1 (126.2) | 127.4 (127.6) |
| FePc b | 1.935 (1.927) | 1.393 (1.378) | 1.321 (1.322) | 126.4 (126.3) | 127.3 (127.8) |
| VPc | CrPc | MnPc | FePc | |
|---|---|---|---|---|
| a1g | 100.9 | 106.7 | 103.8 | 0.0 |
| b1g | −86.8 | −85.9 | −26.8 | 0.0 |
| b2g | 81.6 | 99.6 | 86.2 | 0.0 |
| eg | −16.7 | −55.0 | −153.9 | 0.0 |
| ΔEorb | 79.0 | 64.8 | 9.3 | 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlotto, S.; Sambi, M.; Sedona, F.; Vittadini, A.; Casarin, M. A Theoretical Study of the Occupied and Unoccupied Electronic Structure of High- and Intermediate-Spin Transition Metal Phthalocyaninato (Pc) Complexes: VPc, CrPc, MnPc, and FePc. Nanomaterials 2021, 11, 54. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010054
Carlotto S, Sambi M, Sedona F, Vittadini A, Casarin M. A Theoretical Study of the Occupied and Unoccupied Electronic Structure of High- and Intermediate-Spin Transition Metal Phthalocyaninato (Pc) Complexes: VPc, CrPc, MnPc, and FePc. Nanomaterials. 2021; 11(1):54. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010054
Chicago/Turabian StyleCarlotto, Silvia, Mauro Sambi, Francesco Sedona, Andrea Vittadini, and Maurizio Casarin. 2021. "A Theoretical Study of the Occupied and Unoccupied Electronic Structure of High- and Intermediate-Spin Transition Metal Phthalocyaninato (Pc) Complexes: VPc, CrPc, MnPc, and FePc" Nanomaterials 11, no. 1: 54. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010054