
Drug Targeting of Inflammatory Bowel Diseases by Biomolecules

 , , , ,
, , , ,  , , , and
, , , and 
Abstract
:1. Introduction to Inflammatory Bowel Diseases (IBD)
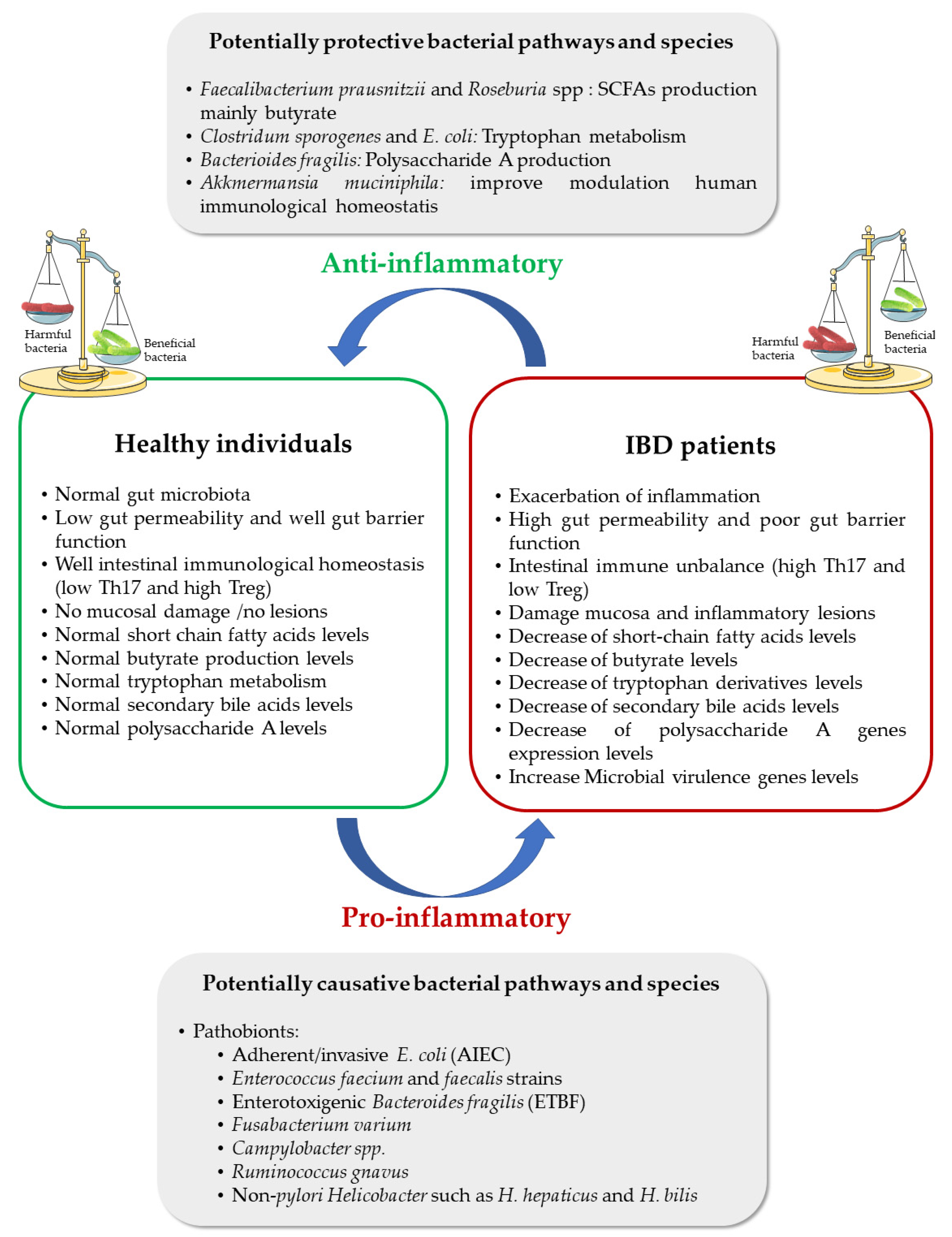
2. Gut Microbiota and IBD Progression
3. IBD Symptoms and Treatment Options
4. Benefits of a Nanomedicine-Based Therapy for IBD
4.1. 5-Aminosalicylic Acid (5-ASA)
4.2. Corticosteroids
4.3. Immunomodulators
4.4. RNA Therapeutic Strategies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- WHO. Noncommunicable Diseases. Available online: https://www.who.int/en/news-room/fact-sheets/detail/noncommunicable-diseases (accessed on 26 June 2021).
- Windsor, J.W.; Kaplan, G.G. Evolving Epidemiology of IBD. Curr. Gastroenterol. Rep. 2019, 21, 40. [Google Scholar] [CrossRef] [PubMed]
- Tenailleau, Q.M.; Lanier, C.; Gower-Rousseau, C.; Cuny, D.; Deram, A.; Occelli, F. Crohn’s disease and environmental contamination: Current challenges and perspectives in exposure evaluation. Environ. Pollut. 2020, 263, 114599. [Google Scholar] [CrossRef] [PubMed]
- SPG. Doença Inflamatória Intestinal—Realidade Atual. Available online: https://www.spg.pt/2016/10/27/doenca-inflamatoria-intestinal-realidade-atual/ (accessed on 26 June 2021).
- Roda, G.; Chien Ng, S.; Kotze, P.G.; Argollo, M.; Panaccione, R.; Spinelli, A.; Kaser, A.; Peyrin-Biroulet, L.; Danese, S. Crohn’s disease. Nat. Rev. Dis. Primers 2020, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Seyed Tabib, N.S.; Madgwick, M.; Sudhakar, P.; Verstockt, B.; Korcsmaros, T.; Vermeire, S. Big data in IBD: Big progress for clinical practice. Gut 2020, 69, 1520–1532. [Google Scholar] [CrossRef]
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Annese, V. Genetics and epigenetics of IBD. Pharmacol. Res. 2020, 159, 104892. [Google Scholar] [CrossRef]
- Younis, N.; Zarif, R.; Mahfouz, R. Inflammatory bowel disease: Between genetics and microbiota. Mol. Biol. Rep. 2020, 47, 3053–3063. [Google Scholar] [CrossRef]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Caminero, A.; Pinto-Sanchez, M.I. Host immune interactions in chronic inflammatory gastrointestinal conditions. Curr. Opin. Gastroenterol. 2020, 36, 479–484. [Google Scholar] [CrossRef]
- Petagna, L.; Antonelli, A.; Ganini, C.; Bellato, V.; Campanelli, M.; Divizia, A.; Efrati, C.; Franceschilli, M.; Guida, A.M.; Ingallinella, S.; et al. Pathophysiology of Crohn’s disease inflammation and recurrence. Biol. Direct 2020, 15, 23. [Google Scholar] [CrossRef]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Tao, W.; Zhu, S. T lymphocytes in the intestinal mucosa: Defense and tolerance. Cell Mol. Immunol. 2019, 16, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Segal, A.W. The role of neutrophils in the pathogenesis of Crohn’s disease. Eur. J. Clin. Invest. 2018, 48, e12983. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Shen, J. Role of environmental factors in the pathogenesis of Crohn’s disease: A critical review. Int. J. Colorectal Dis. 2019, 34, 2023–2034. [Google Scholar] [CrossRef]
- Du, L.; Ha, C. Epidemiology and Pathogenesis of Ulcerative Colitis. Gastroenterol. Clin. North Am. 2020, 49, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.S.E.; Tay, H.L.; Tan, S.H.; Lee, T.H.; Ng, T.M.; Lye, D.C. Gut Microbiota Modulation: Implications for Infection Control and Antimicrobial Stewardship. Adv. Ther. 2020, 37, 4054–4067. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, H.; Liu, Z. Interplay of intestinal microbiota and mucosal immunity in inflammatory bowel disease: A relationship of frenemies. Therap. Adv. Gastroenterol. 2020, 13, 1756284820935188. [Google Scholar] [CrossRef]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Mishima, Y.; Sartor, R.B. Manipulating resident microbiota to enhance regulatory immune function to treat inflammatory bowel diseases. J. Gastroenterol. 2020, 55, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Nagao-Kitamoto, H.; Shreiner, A.B.; Gillilland, M.G.; Kitamoto, S.; Ishii, C.; Hirayama, A.; Kuffa, P.; El-Zaatari, M.; Grasberger, H.; Seekatz, A.M.; et al. Functional Characterization of Inflammatory Bowel Disease-Associated Gut Dysbiosis in Gnotobiotic Mice. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 468–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartor, B. Microbial-host interactions in inflammatory bowel diseases and experimental colitis. Nestle Nutr. Workshop Ser. Pediatr. Program 2009, 64, 121–137. [Google Scholar] [CrossRef]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of gut microbiota in inflammatory bowel disease (IBD): Cause or consequence? IBD treatment targeting the gut microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [Green Version]
- Henson, M.A.; Phalak, P. Microbiota dysbiosis in inflammatory bowel diseases: In silico investigation of the oxygen hypothesis. BMC Syst. Biol. 2017, 11, 145. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; et al. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORγt+ Regulatory T Cells and Exacerbate Colitis in Mice. Immunity 2019, 50, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Caenepeel, C.; Sadat Seyed Tabib, N.; Vieira-Silva, S.; Vermeire, S. Review article: How the intestinal microbiota may reflect disease activity and influence therapeutic outcome in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2020, 52, 1453–1468. [Google Scholar] [CrossRef] [PubMed]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species roseburia hominis and faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Lepage, P.; Hösler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Doré, J.; Raedler, A.; et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 13, 17004. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; Vallarino, C.; Lissoos, T.; Darr, U.; Luo, M. Risk of Infection and Types of Infection among Elderly Patients with Inflammatory Bowel Disease: A Retrospective Database Analysis. Inflamm. Bowel Dis. 2020, 26, 462–468. [Google Scholar] [CrossRef]
- Rees, W.D.; Lorenzo-Leal, A.C.; Steiner, T.S.; Bach, H. Mycobacterium avium subspecies paratuberculosis infects and replicates within human monocyte-derived dendritic cells. Microorganisms 2020, 8, 994. [Google Scholar] [CrossRef]
- Moens, A.; Verstockt, B.; Machiels, K.; Bossuyt, P.; Verdonck, A.; Lagrou, K.; Van Assche, G.; Vermeire, S.; Ferrante, M. Clostridium difficile infection in inflammatory bowel disease: Epidemiology over two decades. Eur. J. Gastroenterol. Hepatol. 2019, 31, 668–673. [Google Scholar] [CrossRef]
- Lin, K.D.; Chiu, G.F.; Waljee, A.K.; Owyang, S.Y.; El-Zaatari, M.; Bishu, S.; Grasberger, H.; Zhang, M.; Wu, D.C.; Kao, J.Y. Effects of Anti–Helicobacter pylori Therapy on Incidence of Autoimmune Diseases, Including Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2019, 17, 1991–1999. [Google Scholar] [CrossRef]
- Nielsen, H.L.; Dalager-Pedersen, M.; Nielsen, H. Risk of inflammatory bowel disease after Campylobacter jejuni and Campylobacter concisus infection: A population-based cohort study. Scand. J. Gastroenterol. 2019, 54, 265–272. [Google Scholar] [CrossRef]
- Alvarez, C.A.; Jones, M.B.; Hambor, J.; Cobb, B.A. Characterization of Polysaccharide A Response Reveals Interferon Responsive Gene Signature and Immunomodulatory Marker Expression. Front. Immunol. 2020, 11, 556813. [Google Scholar] [CrossRef]
- Delmas, J.; Gibold, L.; Faïs, T.; Batista, S.; Leremboure, M.; Sinel, C.; Vazeille, E.; Cattoir, V.; Buisson, A.; Barnich, N.; et al. Metabolic adaptation of adherent-invasive Escherichia coli to exposure to bile salts. Sci. Rep. 2019, 9, 2175. [Google Scholar] [CrossRef]
- de Jong, R.J.; Ohnmacht, C. Defining Dysbiosis in Inflammatory Bowel Disease. Immunity 2019, 50, 8–10. [Google Scholar] [CrossRef] [Green Version]
- Björkqvist, O.; Repsilber, D.; Seifert, M.; Brislawn, C.; Jansson, J.; Engstrand, L.; Rangel, I.; Halfvarson, J. Alterations in the relative abundance of Faecalibacterium prausnitzii correlate with changes in fecal calprotectin in patients with ileal Crohn’s disease: A longitudinal study. Scand. J. Gastroenterol. 2019, 54, 577–585. [Google Scholar] [CrossRef]
- Daliri, E.B.M.; Ofosu, F.K.; Chelliah, R.; Lee, B.H.; Oh, D.H. Health impact and therapeutic manipulation of the gut microbiome. High Throughput 2020, 9, 17. [Google Scholar] [CrossRef]
- Bergmann, H.; Roth, S.; Pechloff, K.; Kiss, E.A.; Kuhn, S.; Heikenwälder, M.; Diefenbach, A.; Greten, F.R.; Ruland, J. Card9-dependent IL-1β regulates IL-22 production from group 3 innate lymphoid cells and promotes colitis-associated cancer. Eur. J. Immunol. 2017, 47, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earley, H.; Lennon, G.; Balfe, Á.; Coffey, J.C.; Winter, D.C.; O’Connell, P.R. The abundance of Akkermansia muciniphila and its relationship with sulphated colonic mucins in health and ulcerative colitis. Sci. Rep. 2019, 9, 15683. [Google Scholar] [CrossRef] [Green Version]
- Plovier, H.; Everard, A.; Druart, C.; Depommier, C.; Van Hul, M.; Geurts, L.; Chilloux, J.; Ottman, N.; Duparc, T.; Lichtenstein, L.; et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 2017, 23, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; Di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J.F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef]
- Mann, E.A.; Saeed, S.A. Gastrointestinal infection as a trigger for inflammatory bowel disease. Curr. Opin. Gastroenterol. 2012, 28, 24–29. [Google Scholar] [CrossRef]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef]
- Leccese, G.; Bibi, A.; Mazza, S.; Facciotti, F.; Caprioli, F.; Landini, P.; Paroni, M. Probiotic Lactobacillus and Bifidobacterium Strains Counteract Adherent-Invasive Escherichia coli (AIEC) Virulence and Hamper IL-23/Th17 Axis in Ulcerative Colitis, but Not in Crohn’s Disease. Cells 2020, 9, 1824. [Google Scholar] [CrossRef]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; McIver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Paccagnini, D.; Vidali, F.; Rosu, V.; Biancheri, P.; Cossu, A.; Zanetti, S.; Corazza, G.R.; Sechi, L.A. Detection of Mycobacterium avium subsp. paratuberculosis (MAP)-specific IS900 DNA and antibodies against MAP peptides and lysate in the blood of Crohn’s disease patients. Inflamm. Bowel Dis. 2011, 17, 1254–1255. [Google Scholar] [CrossRef] [Green Version]
- Juste, R.A.; Elguezabal, N.; Garrido, J.M.; Pavon, A.; Geijo, M.V.; Sevilla, I.; Cabriada, J.L.; Tejada, A.; Garcia-Campos, F.; Casado, R.; et al. On the prevalence of M. avium subspecies paratuberculosis DNA in the blood of healthy individuals and patients with inflammatory bowel disease. PLoS ONE 2008, 3, e2537. [Google Scholar] [CrossRef]
- Navaneethan, U.; Venkatesh, P.G.K.; Shen, B. Clostridium difficile infection and inflammatory bowel disease: Understanding the evolving relationship. World J. Gastroenterol. 2010, 16, 4892–4904. [Google Scholar] [CrossRef]
- Shoaei, P.; Shojaei, H.; Jalali, M.; Khorvash, F.; Hosseini, S.M.; Ataei, B.; Vakili, B.; Ebrahimi, F.; Tavakoli, H.; Esfandiari, Z.; et al. Clostridium difficile isolated from faecal samples in patients with ulcerative colitis. BMC Infect. Dis. 2019, 19, 361. [Google Scholar] [CrossRef]
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory bowel disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.; Zuo, T.; Ho, M.; Chan, F.K.L.; Chan, P.K.S.; Ng, S.C. Review article: Fungal alterations in inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2019, 50, 1159–1171. [Google Scholar] [CrossRef]
- Hoarau, G.; Mukherjee, P.K.; Gower-Rousseau, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and mycobiome interactions underscore microbial dysbiosis in familial Crohn’s disease. mBio 2016, 7, e01250-16. [Google Scholar] [CrossRef] [Green Version]
- Dal Buono, A.; Roda, G.; Argollo, M.; Zacharopoulou, E.; Peyrin-Biroulet, L.; Danese, S. Treat to target or ‘treat to clear’ in inflammatory bowel diseases: One step further? Expert Rev. Gastroenterol. Hepatol. 2020, 14, 807–817. [Google Scholar] [CrossRef]
- Guedj, K.; Abitbol, Y.; Cazals-Hatem, D.; Morvan, M.; Maggiori, L.; Panis, Y.; Bouhnik, Y.; Caligiuri, G.; Corcos, O.; Nicoletti, A. Adipocytes orchestrate the formation of tertiary lymphoid organs in the creeping fat of Crohn’s disease affected mesentery. J. Autoimmun. 2019, 103, 102281. [Google Scholar] [CrossRef]
- Van Kruiningen, H.J. What the early pathologists got wrong, and right, about the pathology of Crohn’s disease: A historical perspective. APMIS 2020, 128, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Kaenkumchorn, T.; Wahbeh, G. Ulcerative Colitis: Making the Diagnosis. Gastroenterol. Clin. North Am. 2020, 49, 655–669. [Google Scholar] [CrossRef]
- Berg, D.R.; Colombel, J.F.; Ungaro, R. The role of early biologic therapy in inflammatory bowel disease. Inflamm. Bowel Dis. 2019, 25, 1905. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Goggolidou, P. Ulcerative colitis: Understanding its cellular pathology could provide insights into novel therapies. J. Inflamm. (Lond) 2020, 17, 15. [Google Scholar] [CrossRef] [Green Version]
- Lo, B.; Zhao, M.; Vind, I.; Burisch, J. The Risk of Extraintestinal Cancer in Inflammatory Bowel Disease: A Systematic Review and Meta-analysis of Population-based Cohort Studies. Clin. Gastroenterol. Hepatol. 2021, 19, 1117–1138.e1119. [Google Scholar] [CrossRef]
- Chang, M.; Chang, L.; Chang, H.M.; Chang, F. Intestinal and Extraintestinal Cancers Associated With Inflammatory Bowel Disease. Clin. Colorectal Cancer 2018, 17, e29–e37. [Google Scholar] [CrossRef] [Green Version]
- Román, A.L.S.; Muñoz, F. Comorbidity in inflammatory bowel disease. World J. Gastroenterol. 2011, 17, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Argollo, M.; Gilardi, D.; Peyrin-Biroulet, C.; Chabot, J.F.; Peyrin-Biroulet, L.; Danese, S. Comorbidities in inflammatory bowel disease: A call for action. Lancet Gastroenterol. Hepatol. 2019, 4, 643–654. [Google Scholar] [CrossRef]
- Tinsley, A.; Navabi, S.; Williams, E.D.; Liu, G.; Kong, L.; Coates, M.D.; Clarke, K. Increased Risk of Influenza and Influenza-Related Complications among 140,480 Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Romano, C.; Esposito, S.; Ferrara, R.; Cuomo, G. Choosing the most appropriate biologic therapy for Crohn’s disease according to concomitant extra-intestinal manifestations, comorbidities, or physiologic conditions. Expert Opin. Biol. Ther. 2020, 20, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, P.; Singh, K. Therapeutic applications of probiotics in ulcerative colitis: An updated review. PharmaNutrition 2020, 13, 100194. [Google Scholar] [CrossRef]
- Mowat, C.; Cole, A.; Windsor, A.; Ahmad, T.; Arnott, I.; Driscoll, R.; Mitton, S.; Orchard, T.; Rutter, M.; Younge, L.; et al. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011, 60, 571–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, K.; Feuerstein, J.D. New developments in ulcerative colitis: Latest evidence on management, treatment, and maintenance. Drugs Context 2019, 8, 212572. [Google Scholar] [CrossRef]
- Katz, S.; Liu, Y. Challenges in the Management of Inflammatory Bowel Disease. In Geriatric Gastroenterology; Pitchumoni, C.S., Dharmarajan, T., Eds.; Springer: Cham, Switzerland, 2020; pp. 1–16. [Google Scholar] [CrossRef]
- Magro, F.; Cordeiro, G.; Dias, A.M.; Estevinho, M.M. Inflammatory Bowel Disease—Non-biological treatment. Pharmacol. Res. 2020, 160, 105075. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 2019, 68, s1–s106. [Google Scholar] [CrossRef] [Green Version]
- Privitera, G.; Pugliese, D.; Onali, S.; Petito, V.; Scaldaferri, F.; Gasbarrini, A.; Danese, S.; Armuzzi, A. Combination therapy in inflammatory bowel disease—From traditional immunosuppressors towards the new paradigm of dual targeted therapy. Autoimmun. Rev. 2021, 20. [Google Scholar] [CrossRef] [PubMed]
- Hazel, K.; O’Connor, A. Emerging treatments for inflammatory bowel disease. Ther. Adv. Chronic Dis. 2020, 11, 2040622319899297. [Google Scholar] [CrossRef]
- Mao, E.J.; Hazlewood, G.S.; Kaplan, G.G.; Peyrin-Biroulet, L.; Ananthakrishnan, A.N. Systematic review with meta-analysis: Comparative efficacy of immunosuppressants and biologics for reducing hospitalisation and surgery in Crohn’s disease and ulcerative colitis. Aliment Pharmacol. Ther. 2017, 45, 3–13. [Google Scholar] [CrossRef]
- Sandborn, W.; Feagan, B.; Danese, S.; O’ Brien, C.; Ott, E.; Marano, C.; Baker, T.; Zhou, Y.; Volger, S.; Tikhonov, I.; et al. Safety of Ustekinumab in Inflammatory Bowel Disease: Pooled Safety Analysis of Results from Phase 2/3 Studies. Inflamm. Bowel Dis. 2021, 27, 994–1007. [Google Scholar] [CrossRef]
- Côté-Daigneault, J.; Bouin, M.; Lahaie, R.; Colombel, J.F.; Poitras, P. Biologics in inflammatory bowel disease: What are the data? United Eur. Gastroenterol. J. 2015, 3, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Kayal, M.; Shah, S. Ulcerative Colitis: Current and Emerging Treatment Strategies. J. Clin. Med. 2019, 9, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivaji, U.N.; Sharratt, C.L.; Thomas, T.; Smith, S.C.L.; Iacucci, M.; Moran, G.W.; Ghosh, S.; Bhala, N. Review article: Managing the adverse events caused by anti-TNF therapy in inflammatory bowel disease. Aliment Pharmacol. Ther. 2019, 49, 664–680. [Google Scholar] [CrossRef] [Green Version]
- Dotan, I.; Allez, M.; Danese, S.; Keir, M.; Tole, S.; McBride, J. The role of integrins in the pathogenesis of inflammatory bowel disease: Approved and investigational anti-integrin therapies. Med. Res. Rev. 2020, 40, 245–262. [Google Scholar] [CrossRef] [Green Version]
- Takatsu, N.; Hisabe, T.; Higashi, D.; Ueki, T.; Matsui, T. Vedolizumab in the Treatment of Ulcerative Colitis: An Evidence-Based Review of Safety, Efficacy, and Place of Therapy. Core Evid. 2020, 15, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiot, A.; Filippi, J.; Abitbol, V.; Cadiot, G.; Laharie, D.; Serrero, M.; Altwegg, R.; Bouhnik, Y.; Peyrin-Biroulet, L.; Gilletta, C.; et al. Effectiveness and safety of ustekinumab induction therapy for 103 patients with ulcerative colitis: A GETAID multicentre real-world cohort study. Aliment Pharmacol. Ther. 2020, 51, 1039–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Baumgart, D.C. Calcineurin inhibitors in ulcerative colitis. In Crohn’s Disease and Ulcerative Colitis; Baumgart, D.C., Ed.; Springer: Cham, Switzerland, 2017; pp. 421–428. [Google Scholar] [CrossRef]
- Rogler, G. Efficacy of JAK inhibitors in Crohn’s Disease. J. Crohns Colitis 2020, 14, S746–S754. [Google Scholar] [CrossRef]
- Harris, C.; Cummings, J.R.F. JAK1 inhibition and inflammatory bowel disease. Rheumatology 2021, 60, ii45–ii51. [Google Scholar] [CrossRef]
- Schmidt, C.; Grunert, P.C.; Stallmach, A. An Update for Pharmacologists on New Treatment Options for Inflammatory Bowel Disease: The Clinicians’ Perspective. Front. Pharmacol. 2021, 12, 655054. [Google Scholar] [CrossRef]
- Misselwitz, B.; Juillerat, P.; Sulz, M.C.; Siegmund, B.; Brand, S. Emerging Treatment Options in Inflammatory Bowel Disease: Janus Kinases, Stem Cells, and More. Diggestion 2020, 101, 69–82. [Google Scholar] [CrossRef]
- Chen, W.; Chen, H.; Fu, S.; Lin, X.; Zheng, Z.; Zhang, J. Microbiome characterization and re-design by biologic agents for inflammatory bowel disease insights. Bioprocess Biosyst. Eng. 2021, 44, 929–939. [Google Scholar] [CrossRef]
- Van Praag, E.M.; Buskens, C.J.; Hompes, R.; Bemelman, W.A. Surgical management of Crohn’s disease: A state of the art review. Int. J. Colorectal Dis. 2021, 36, 1133–1145. [Google Scholar] [CrossRef]
- Carrière, J.; Darfeuille-Michaud, A.; Nguyen, H.T.T. Infectious etiopathogenesis of Crohn’s disease. World J. Gastroenterol. 2014, 20, 12102–12117. [Google Scholar] [CrossRef]
- Nitzan, O.; Elias, M.; Peretz, A.; Saliba, W. Role of antibiotics for treatment of inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhao, W.; Lan, P.; Mou, X. The microbiome in inflammatory bowel diseases: From pathogenesis to therapy. Protein Cell 2021, 12, 331–345. [Google Scholar] [CrossRef]
- Galtier, M.; De Sordi, L.; Sivignon, A.; de Vallée, A.; Maura, D.; Neut, C.; Rahmouni, O.; Wannerberger, K.; Darfeuille-Michaud, A.; Desreumaux, P.; et al. Bacteriophages targeting adherent invasive Escherichia coli strains as a promising new treatment for Crohn’s disease. J. Crohns Colitis 2017, 11, 840–847. [Google Scholar] [CrossRef] [Green Version]
- Glassner, K.L.; Abraham, B.P.; Quigley, E.M.M. The microbiome and inflammatory bowel disease. J. Allergy Clin. Immunol. 2020, 145, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, X.; Wu, W.; Yang, L.; Lv, L.; Wang, Q.; Li, Y.; Ye, J.; Fang, D.; Wu, J.; Jiang, X.; et al. Administration of Akkermansia muciniphila Ameliorates Dextran Sulfate Sodium-Induced Ulcerative Colitis in Mice. Front. Microbiol. 2019, 10, 2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Tang, L.; Feng, Y.; Zhao, S.; Han, M.; Zhang, C.; Yuan, G.; Zhu, J.; Cao, S.; Wu, Q.; et al. A purified membrane protein from Akkermansia muciniphila or the pasteurised bacterium blunts colitis associated tumourigenesis by modulation of CD8+ T cells in mice. Gut 2020, 69, 1988–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akram, W.; Garud, N.; Joshi, R. Role of inulin as prebiotics on inflammatory bowel disease. Drug Discov. Ther. 2019, 13, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, T.; Wong, S.H.; Cheung, C.P.; Lam, K.; Lui, R.; Cheung, K.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Wu, J.C.Y.; et al. Gut fungal dysbiosis correlates with reduced efficacy of fecal microbiota transplantation in Clostridium difficile infection. Nat. Commun. 2018, 9, 3663. [Google Scholar] [CrossRef]
- Collij, V.; Klaassen, M.A.Y.; Weersma, R.K.; Vila, A.V. Gut microbiota in inflammatory bowel diseases: Moving from basic science to clinical applications. Hum. Genet. 2021, 140, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Plichta, D.R.; Graham, D.B.; Subramanian, S.; Xavier, R.J. Therapeutic Opportunities in Inflammatory Bowel Disease: Mechanistic Dissection of Host-Microbiome Relationships. Cell 2019, 178, 1041–1056. [Google Scholar] [CrossRef]
- Macaluso, F.S.; Maida, M.; Grova, M.; Crispino, F.; Teresi, G.; Orlando, A.; Orlando, A. Head-to-head comparison of biological drugs for inflammatory bowel disease: From randomized controlled trials to real-world experience. Therap. Adv. Gastroenterol. 2021, 14, 17562848211010668. [Google Scholar] [CrossRef]
- Liu, W.; Dong, Z.; Liu, K.; Lu, Y.; Wu, W.; Qi, J.; Chen, Z. Targeting strategies of oral nano-delivery systems for treating inflammatory bowel disease. Int. J. Pharm. 2021, 600, 120461. [Google Scholar] [CrossRef]
- Antunes, J.C.; Benarroch, L.; Moraes, F.C.; Juenet, M.; Gross, M.S.; Aubart, M.; Boileau, C.; Caligiuri, G.; Nicoletti, A.; Ollivier, V.; et al. Core-Shell Polymer-Based Nanoparticles Deliver miR-155-5p to Endothelial Cells. Mol. Ther. Nucleic Acids 2019, 17, 210–222. [Google Scholar] [CrossRef] [Green Version]
- Antunes, J.C.; Gonçalves, R.M.; Barbosa, M.A. Chitosan/poly(γ-glutamic acid) polyelectrolyte complexes: From self-assembly to application in biomolecules delivery and regenerative medicine. Res. Rev. J. Mater. Sci. 2016, 4, 12–36. [Google Scholar] [CrossRef]
- Moraes, F.C.; Marcelo Forero Ramirez, L.; Aid, R.; Benadda, S.; Maire, M.; Chauvierre, C.; Antunes, J.C.; Chaubet, F.; Letourneur, D. P-selectin targeting polysaccharide-based nanogels for miRNA delivery. Int. J. Pharm. 2021, 597, 120302. [Google Scholar] [CrossRef]
- Mele, E. Electrospinning of essential oils. Polymers 2020, 12, 908. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Seyedian, S.S.; Nokhostin, F.; Malamir, M.D. A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease. J. Med. Life 2019, 12, 113–122. [Google Scholar] [CrossRef]
- Date, A.A.; Hanes, J.; Ensign, L.M. Nanoparticles for oral delivery: Design, evaluation and state-of-the-art. J. Control. Release 2016, 240, 504–526. [Google Scholar] [CrossRef] [Green Version]
- dos Santos, A.M.; Carvalho, S.G.; Meneguin, A.B.; Sábio, R.M.; Gremião, M.P.D.; Chorilli, M. Oral delivery of micro/nanoparticulate systems based on natural polysaccharides for intestinal diseases therapy: Challenges, advances and future perspectives. J. Control. Release 2021, 334, 353–366. [Google Scholar] [CrossRef]
- Lautenschläger, C.; Schmidt, C.; Fischer, D.; Stallmach, A. Drug delivery strategies in the therapy of inflammatory bowel disease. Adv. Drug Deliv. Rev. 2014, 71, 58–76. [Google Scholar] [CrossRef]
- Hua, S.; Marks, E.; Schneider, J.J.; Keely, S. Advances in oral nano-delivery systems for colon targeted drug delivery in inflammatory bowel disease: Selective targeting to diseased versus healthy tissue. Nanomedicine 2015, 11, 1117–1132. [Google Scholar] [CrossRef] [Green Version]
- Barani, M.; Rahdar, A.; Sargazi, S.; Amiri, M.S.; Sharma, P.K.; Bhalla, N. Nanotechnology for inflammatory bowel disease management: Detection, imaging and treatment. Sens. Bio-Sensing Res. 2021, 32, 100417. [Google Scholar] [CrossRef]
- Costa-Lima, S.A.; Reis, S. Nanotechnological Approaches in Drug Absorption through Skin Topical Delivery. In Nanoparticles in the Life Sciences and Biomedicine; Neves, A.R., Reis, S., Eds.; Jenny Stanford Publishing: Boca Raton, FL, USA, 2018. [Google Scholar]
- Nakkala, J.R.; Li, Z.; Ahmad, W.; Wang, K.; Gao, C. Immunomodulatory biomaterials and their application in therapies for chronic inflammation-related diseases. Acta Biomater. 2021, 123, 1–30. [Google Scholar] [CrossRef]
- Nedelcu, A.; Mosteanu, O.; Pop, T.; Mocan, T.; Mocan, L. Recent advances in nanoparticle-mediated treatment of inflammatory bowel diseases. Appl. Sci. 2021, 11, 438. [Google Scholar] [CrossRef]
- Zhang, L.; Gu, F.X.; Chan, J.M.; Wang, A.Z.; Langer, R.S.; Farokhzad, O.C. Nanoparticles in medicine: Therapeutic applications and developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [Google Scholar] [CrossRef]
- Kulkarni, A.A.; Rao, P.S. Synthesis of polymeric nanomaterials for biomedical applications. In Nanomaterials in Tissue Engineering; Gaharwar, A.K., Sant, S., Hancock, M.J., Hacking, S.A., Eds.; Woodhead Publishing: Sawston, UK, 2013; pp. 27–63. [Google Scholar] [CrossRef]
- Cesar, A.L.A.; Abrantes, F.A.; Farah, L.; Castilho, R.O.; Cardoso, V.; Fernandes, S.O.; Araújo, I.D.; Faraco, A.A.G. New mesalamine polymeric conjugate for controlled release: Preparation, characterization and biodistribution study. Eur. J. Pharm. Sci. 2018, 111, 57–64. [Google Scholar] [CrossRef]
- Malviya, T.; Joshi, S.; Dwivedi, L.M.; Baranwal, K.; Shehala; Pandey, A. K.; Singh, V. Synthesis of Aloevera/Acrylonitrile based Nanoparticles for targeted drug delivery of 5-Aminosalicylic acid. Int. J. Biol. Macromol. 2018, 106, 930–939. [Google Scholar] [CrossRef]
- Singh, V.; Joshi, S.; Malviya, T. Carboxymethyl cellulose-rosin gum hybrid nanoparticles: An efficient drug carrier. Int. J. Biol. Macromol. 2018, 112, 390–398. [Google Scholar] [CrossRef]
- Bahadori, F.; Akinan, B.S.; Akyil, S.; Eroglu, M.S. Synthesis and engineering of sodium alginate/inulin core-shell nano-hydrogels for controlled-release oral delivery of 5-ASA. Org. Commun. 2019, 12, 132–142. [Google Scholar] [CrossRef]
- Nalinbenjapun, S.; Ovatlarnporn, C. Chitosan-5-aminosalicylic acid conjugates for colon-specific drug delivery: Methods of preparation and in vitro evaluations. J. Drug Deliv. Sci. Technol. 2020, 57, 101397. [Google Scholar] [CrossRef]
- Alagozlu, H.; Gorgul, A.; Bilgihan, A.; Tuncer, C.; Unal, S. Increased plasma levels of advanced oxidation protein products (AOPP) as a marker for oxidative stress in patients with active ulcerative colitis. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 80–85. [Google Scholar] [CrossRef]
- Iwao, Y.; Tomiguchi, I.; Domura, A.; Mantaira, Y.; Minami, A.; Suzuki, T.; Ikawa, T.; Kimura, S.I.; Itai, S. Inflamed site-specific drug delivery system based on the interaction of human serum albumin nanoparticles with myeloperoxidase in a murine model of experimental colitis. Eur. J. Pharm. Biopharm. 2018, 125, 141–147. [Google Scholar] [CrossRef]
- Ali, H.; Weigmann, B.; Neurath, M.F.; Collnot, E.M.; Windbergs, M.; Lehr, C.M. Budesonide loaded nanoparticles with pH-sensitive coating for improved mucosal targeting in mouse models of inflammatory bowel diseases. J. Control. Release 2014, 183, 167–177. [Google Scholar] [CrossRef]
- Sinhmar, G.K.; Shah, N.N.; Rawal, S.U.; Chokshi, N.V.; Khatri, H.N.; Patel, B.M.; Patel, M.M. Surface engineered lipid nanoparticle-mediated site-specific drug delivery system for the treatment of inflammatory bowel disease. Artif. Cells Nanomed. Biotechnol. 2018, 46, 565–578. [Google Scholar] [CrossRef]
- Qelliny, M.R.; Aly, U.F.; Elgarhy, O.H.; Khaled, K.A. Budesonide-Loaded Eudragit S 100 Nanocapsules for the Treatment of Acetic Acid-Induced Colitis in Animal Model. AAPS PharmSciTech 2019, 20, 237. [Google Scholar] [CrossRef]
- Sun, Q.; Luan, L.; Arif, M.; Li, J.; Dong, Q.J.; Gao, Y.; Chi, Z.; Liu, C.G. Redox-sensitive nanoparticles based on 4-aminothiophenol-carboxymethyl inulin conjugate for budesonide delivery in inflammatory bowel diseases. Carbohydr. Polym. 2018, 189, 352–359. [Google Scholar] [CrossRef]
- Date, A.A.; Halpert, G.; Babu, T.; Ortiz, J.; Kanvinde, P.; Dimitrion, P.; Narayan, J.; Zierden, H.; Betageri, K.; Musmanno, O.; et al. Mucus-penetrating budesonide nanosuspension enema for local treatment of inflammatory bowel disease. Biomaterials 2018, 185, 97–105. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Liu, Z.; Kerdsakundee, N.; Zhang, M.; Zhang, F.; Liu, X.; Bauleth-Ramos, T.; Lian, W.; Mäkilä, E.; et al. Hierarchical structured and programmed vehicles deliver drugs locally to inflamed sites of intestine. Biomaterials 2018, 185, 322–332. [Google Scholar] [CrossRef]
- Lee, A.; De Mei, C.; Fereira, M.; Marotta, R.; Yoon, H.Y.; Kim, K.; Kwon, I.C.; Decuzzi, P. Dexamethasone-loaded polymeric nanoconstructs for monitoring and treating inflammatory bowel disease. Theranostics 2017, 7, 3653–3666. [Google Scholar] [CrossRef]
- Wang, X.; Yan, J.J.; Wang, L.; Pan, D.; Yang, R.; Xu, Y.P.; Sheng, J.; Huang, Q.; Zhao, H.; Yang, M. Rational design of polyphenol-poloxamer nanovesicles for targeting inflammatory bowel disease therapy. Chem. Mater. 2018, 30, 4073–4080. [Google Scholar] [CrossRef]
- Mukhtar, M.; Zesshan, M.; Khan, S.; Shahnaz, G.; Khan, S.A.; Sarwar, H.S.; Pasha, R.A.; Ali, H. Fabrication and optimization of pH-sensitive mannose-anchored nano-vehicle as a promising approach for macrophage uptake. Appl. Nanosci. 2020, 10, 4013–4027. [Google Scholar] [CrossRef]
- Wang, B.; Zhuang, X.; Deng, Z.B.; Jiang, H.; Mu, J.; Wang, Q.; Xiang, X.; Guo, H.; Zhang, L.; Dryden, G.; et al. Targeted drug delivery to intestinal macrophages by bioactive nanovesicles released from grapefruit. Mol. Ther. 2014, 22, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Naeem, M.; Bae, J.; Oshi, M.A.; Kim, M.S.; Moon, H.R.; Lee, B.L.; Im, E.; Jung, Y.; Yoo, J.W. Colon-targeted delivery of cyclosporine a using dual-functional eudragit® FS30D/PLGA nanoparticles ameliorates murine experimental colitis. Int. J. Nanomedicine 2018, 13, 1225–1240. [Google Scholar] [CrossRef] [Green Version]
- Courthion, H.; Mugnier, T.; Rousseaux, C.; Möller, M.; Gurny, R.; Gabriel, D. Self-assembling polymeric nanocarriers to target inflammatory lesions in ulcerative colitis. J. Control. Release 2018, 275, 32–39. [Google Scholar] [CrossRef]
- Melero, A.; Draheim, C.; Hansen, S.; Giner, E.; Carreras, J.J.; Talens-Visconti, R.; Garrigues, T.M.; Peris, J.E.; Recio, M.C.; Giner, R.; et al. Targeted delivery of Cyclosporine A by polymeric nanocarriers improves the therapy of inflammatory bowel disease in a relevant mouse model. Eur. J. Pharm. Biopharm. 2017, 119, 361–371. [Google Scholar] [CrossRef]
- Laroui, H.; Dalmasso, G.; Nguyen, H.T.T.; Yan, Y.; Sitaraman, S.V.; Merlin, D. Drug-Loaded Nanoparticles Targeted to the Colon with Polysaccharide Hydrogel Reduce Colitis in a Mouse Model. Gastroenterology 2010, 138, 843–853.e842. [Google Scholar] [CrossRef] [PubMed]
- Laroui, H.; Geem, D.; Xiao, B.; Viennois, E.; Rakhya, P.; Denning, T.; Merlin, D. Targeting intestinal inflammation with CD98 siRNA/PEI-loaded nanoparticles. Mol. Ther. 2014, 22, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Xu, Z.; Viennois, E.; Zhang, Y.; Zhang, Z.; Zhang, M.; Han, M.K.; Kang, Y.; Merlin, D. Orally Targeted Delivery of Tripeptide KPV via Hyaluronic Acid-Functionalized Nanoparticles Efficiently Alleviates Ulcerative Colitis. Mol. Ther. 2017, 25, 1628–1640. [Google Scholar] [CrossRef] [Green Version]
- Frede, A.; Neuhaus, B.; Klopfleisch, R.; Walker, C.; Buer, J.; Müller, W.; Epple, M.; Westendorf, A.M. Colonic gene silencing using siRNA-loaded calcium phosphate/PLGA nanoparticles ameliorates intestinal inflammation in vivo. J. Control. Release 2016, 222, 86–96. [Google Scholar] [CrossRef]
- Xiao, B.; Chen, Q.; Zhang, Z.; Wang, L.; Kang, Y.; Denning, T.; Merlin, D. TNFα gene silencing mediated by orally targeted nanoparticles combined with interleukin-22 for synergistic combination therapy of ulcerative colitis. J. Control. Release 2018, 287, 235–246. [Google Scholar] [CrossRef]
- Duan, B.; Li, M.; Sun, Y.; Zou, S.; Xu, X. Orally Delivered Antisense Oligodeoxyribonucleotides of TNF-α via Polysaccharide-Based Nanocomposites Targeting Intestinal Inflammation. Adv. Healthc. Mater. 2019, 8, e1801389. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, J.; Li, Y.; Zhao, R.; Du, S.; Lv, C.; Wu, W.; Liu, R.; Sheng, X.; Song, Y.; et al. MicroRNA-31 Reduces Inflammatory Signaling and Promotes Regeneration in Colon Epithelium, and Delivery of Mimics in Microspheres Reduces Colitis in Mice. Gastroenterology 2019, 156, 2281–2296.e2286. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, C.; Yin, C. Galactosylated trimethyl chitosan-cysteine nanoparticles loaded with Map4k4 siRNA for targeting activated macrophages. Biomaterials 2013, 34, 3667–3677. [Google Scholar] [CrossRef]
- Anitha, A.; Rejinold, S.N.; Bumgardner, J.D.; Nair, S.V.; Jayakumar, R. Approaches for Functional Modification or Cross-Linking of Chitosan. In Chitosan-Based Systems for Biopharmaceuticals: Delivery, Targeting and Polymer Therapeutics; John Wiley and Sons: Hoboken, NJ, USA, 2012; pp. 107–124. [Google Scholar] [CrossRef] [Green Version]
- Kaolaor, A.; Phunpee, S.; Ruktanonchai, U.R.; Suwantong, O. Effects of β-cyclodextrin complexation of curcumin and quaternization of chitosan on the properties of the blend films for use as wound dressings. J. Polym. Res. 2019, 26, 43. [Google Scholar] [CrossRef]
- Moraes, F.C.; Antunes, J.C.; Forero Ramirez, L.M.; Aprile, P.; Franck, G.; Chauvierre, C.; Chaubet, F.; Letourneur, D. Synthesis of cationic quaternized pullulan derivatives for miRNA delivery. Int. J. Pharm. 2020, 577, 119041. [Google Scholar] [CrossRef]
- Xu, X.; Yang, W.; Liang, Q.; Shi, Y.; Zhang, W.; Wang, X.; Meng, F.; Zhong, Z.; Yin, L. Efficient and targeted drug/siRNA co-delivery mediated by reversibly crosslinked polymersomes toward anti-inflammatory treatment of ulcerative colitis (UC). Nano Res. 2019, 12, 659–667. [Google Scholar] [CrossRef]
- Antunes, J.C.; Domingues, J.M.; Miranda, C.S.; Silva, A.F.G.; Homem, N.C.; Amorim, M.T.P.; Felgueiras, H.P. Bioactivity of chitosan-based particles loaded with plant-derived extracts for biomedical applications: Emphasis on antimicrobial fiber-based systems. Mar. Drugs 2021, 19, 359. [Google Scholar] [CrossRef]
- Maronek, M.; Link, R.; Ambro, L.; Gardlik, R. Phages and Their Role in Gastrointestinal Disease: Focus on Inflammatory Bowel Disease. Cells 2020, 9, 1013. [Google Scholar] [CrossRef] [Green Version]
- Giron, F.; Pastó, A.; Tasciotti, E.; Abraham, B. Nanotechnology in the Treatment of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 1871–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, R.; Neves, J.D.; Sarmento, B. Nanoparticles for the regulation of intestinal inflammation: Opportunities and challenges. Nanomedicine 2019, 14, 2631–2644. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cai, J.; Chen, H.; Zhong, Q.; Hou, Y.; Chen, W.; Chen, W. Antibacterial activity and mechanism of linalool against Pseudomonas aeruginosa. Microb. Pathog. 2020, 141, 103980. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.; Escher, J.; Hébuterne, X.; Kłęk, S.; Krznaric, Z.; Schneider, S.; Shamir, R.; Stardelova, K.; Wierdsma, N.; Wiskin, A.; et al. ESPEN practical guideline: Clinical Nutrition in inflammatory bowel disease. Clin. Nutr. 2020, 39, 632–653. [Google Scholar] [CrossRef] [Green Version]
- Green, N.; Miller, T.; Suskind, D.; Lee, D. A Review of Dietary Therapy for IBD and a Vision for the Future. Nutrients 2019, 11, 947. [Google Scholar] [CrossRef] [Green Version]
- Dubinsky, V.; Reshef, L.; Bar, N.; Keizer, D.; Golan, N.; Rabinowitz, K.; Godny, L.; Yadgar, K.; Zonensain, K.; Tulchinsky, H.; et al. Predominantly Antibiotic-resistant Intestinal Microbiome Persists in Patients With Pouchitis Who Respond to Antibiotic Therapy. Gastroenterology 2020, 158, 610–624.e613. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Predicting treatment |
| |
| Diagnostic tool |
| |
| Treatment | Drug from Bugs |
|
| Bugs as Drug |
| |
| Drug for Bug |
| |
| Decolonization |
| |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antunes, J.C.; Seabra, C.L.; Domingues, J.M.; Teixeira, M.O.; Nunes, C.; Costa-Lima, S.A.; Homem, N.C.; Reis, S.; Amorim, M.T.P.; Felgueiras, H.P. Drug Targeting of Inflammatory Bowel Diseases by Biomolecules. Nanomaterials 2021, 11, 2035. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11082035
Antunes JC, Seabra CL, Domingues JM, Teixeira MO, Nunes C, Costa-Lima SA, Homem NC, Reis S, Amorim MTP, Felgueiras HP. Drug Targeting of Inflammatory Bowel Diseases by Biomolecules. Nanomaterials. 2021; 11(8):2035. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11082035
Chicago/Turabian StyleAntunes, Joana Costa, Catarina Leal Seabra, Joana Margarida Domingues, Marta Oliveira Teixeira, Cláudia Nunes, Sofia Antunes Costa-Lima, Natália Cândido Homem, Salette Reis, Maria Teresa Pessoa Amorim, and Helena Prado Felgueiras. 2021. "Drug Targeting of Inflammatory Bowel Diseases by Biomolecules" Nanomaterials 11, no. 8: 2035. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11082035