
Electronic Properties of Carbon Nanobelts Predicted by Thermally-Assisted-Occupation DFT



1
Department of Physics, National Taiwan University, Taipei 10617, Taiwan
2
Center for Theoretical Physics and Center for Quantum Science and Engineering, National Taiwan University, Taipei 10617, Taiwan
3
Physics Division, National Center for Theoretical Sciences, Taipei 10617, Taiwan
*
Author to whom correspondence should be addressed.
Nanomaterials 2021, 11(9), 2224; https://0-doi-org.brum.beds.ac.uk/10.3390/nano11092224
Submission received: 2 August 2021
/
Revised: 23 August 2021
/
Accepted: 26 August 2021
/
Published: 29 August 2021
(This article belongs to the Special Issue Theoretical Calculation and Molecular Modeling of Nanomaterials)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Accurate prediction of properties of large-scale multi-reference (MR) electronic systems remains difficult for traditional computational methods (e.g., the Hartree–Fock theory and Kohn–Sham density functional theory (DFT)). Recently, thermally-assisted-occupation (TAO)-DFT has been demonstrated to offer reliable description of electronic properties of various large-scale MR electronic systems. Consequently, in this work, TAO-DFT is used to unlock the electronic properties associated with C-Belt[n] (i.e., the carbon nanobelts containing n fused 12-membered carbon rings). Our calculations show that for all the system sizes reported (n = 4–24), C-Belt[n] have singlet ground states. In general, the larger the size of C-Belt[n], the more pronounced the MR character of ground-state C-Belt[n], as evident from the symmetrized von Neumann entropy and the occupation numbers of active TAO-orbitals. Furthermore, the active TAO-orbitals are delocalized along the circumference of C-Belt[n], as evident from the visualization of active TAO-orbitals.
1. Introduction
Carbon is one of the richest elements in forming a number of allotropes. Carbon forms 0D, 1D, and 2D nanostructures, such as the well-known C fullerenes (buckyballs), carbon nanotubes (CNTs), and graphene [1,2]. It is also possible to form a plethora of hybrid carbon nanostructures by combining these well-known nanostructures with other nanostructures. The diverse nature originates from the -, -, and -hybridized carbon atoms as well as the combinations of different types of hybridization. Owing to the unique and diverse properties, carbon-based nanomaterials have been applied in many fields, including nanotechnology, electronics, optics, biotechnology, and materials science. In addition to these applications, carbon nanostructures have also drawn attention to their beauty, creating playgrounds for testing different phenomena. Moreover, in carbon-based molecular science, polycyclic aromatic hydrocarbons (PAHs), for example, can have astronomical and biological importance as well.
Among a variety of carbon nanostructures, although the C fullerenes, CNTs, and graphene have been well studied, belt-shaped carbon nanomaterials have been relatively under-reported [3,4,5,6,7,8,9,10,11]. In fact, belt-shaped carbon nanomaterials can be promising because of their important role in supramolecular chemistry. The structure of belt-shaped carbon molecules may offer new possibilities for the synthesis of chemical compounds with various functional groups. Moreover, owing to the deformation of typical - or -type orbitals, belt-shaped carbon molecules (i.e., unconventional -electron systems) can have fascinating properties for some applications (e.g., optoelectronic applications).
Over the past few years, there has been a renewed interest in synthesizing and applying belt-shaped carbon molecules, where the belt structure is formed with fused benzene rings [4,5,11]. Belt-shaped carbon molecules include [n]cycloparaphenylene ([n]CPP) in which the benzene rings are connected by covalent bonds [4,5], belt[n]arene (also called n-cyclacene [7]) where the benzene rings are fused in a linear fashion, a carbon nanobelt containing the benzene rings fused in a non-linear fashion, etc. These molecules can act as unique macrocyclic hosts, and can also be used in molecular recognition and self-assembly. For example, belt-shaped carbon molecules (e.g., [12]CPP, which has already been synthesized [4]) can be considered as the building blocks of CNTs.
Armchair hydrocarbon belts (e.g., [n]CPP), zigzag hydrocarbon belts (e.g., belt[n]arene or n-cyclacene), zigzag carbon nanobelts, etc. are the common names used for carbon nanobelts [11]. The majority of carbon nanobelts consist of only benzene rings (i.e., 6-membered carbon rings). As is well known, carbon forms various nanostructures with pentagon (5-membered ring) and hexagon (6-membered ring) motifs. However, various evidences have shown that carbon nanostructures with motifs other than 5- and 6-membered rings can also be found. For example, 4- and 8-membered CNTs [12], buckyballs with motifs other than 6-membered rings [13], etc. have been proposed. In addition, belt[n]arene[n]tropilidenes, consisting of 6- and 7-membered rings, have been realized [14]. Accordingly, it seems possible to design and synthesize the carbon nanobelts with motifs other than 5- and 6-membered rings.
Very recently, there have been many attempts to synthesize and apply carbon nanobelts [4,5,11]. Several belt-shaped carbon molecules, which are entirely made of fused 6-membered carbon rings, have been recently synthesized. These carbon nanobelts can be used as templates or seeds to synthesize structurally well-defined CNTs [15]. Most of the carbon nanobelts are made of fused benzene rings or the benzene rings connected by covalent bonds. The bonding of rings may be linear or sideways, offering numerous possibilities for generating carbon nanobelts with different structures and properties [11,14], and hence unlocking new applications.
Among various carbon nanobelts, in this work, we focus on the carbon nanobelts consisting of n fused 12-membered carbon rings (as inspired by the work of Estrada and Simón-Manso [6]), denoted as C-Belt[n], as illustrated in Figure 1. These carbon nanobelts need some attention to understand their properties and potential applications. As of now, it has been difficult to synthesize C-Belt[n], probably because of the high strain and multi-reference (MR) character (as will be shown and discussed later) associated with these carbon nanobelts. According to the recent KS-DFT (Kohn–Sham density functional theory) [16] calculations on C-Belt[n], these carbon nanobelts (also called ) should be highly stable [6]. However, several important electronic properties (e.g., singlet-triplet gap, MR character, etc.) of C-Belt[n] remain unavailable. Moreover, the main challenge in the reliable prediction of electronic properties of belt-shaped molecules should be due to the MR character associated with these molecules. Based on our previous studies on belt-shaped and closely related molecules, such as cyclacenes [7], cyclic boron nanoribbons [17], and cyclic carbon chains [18], we speculate that C-Belt[n] could also possess MR character. It is well-known that KS-DFT employing the commonly used exchange-correlation energy functionals can lead to enormous errors in the predicted properties of MR electronic systems, due to the unsuccessful incorporation of static correlation into these exchange-correlation energy functionals [19,20]. Consequently, the properties of C-Belt[n] should be explored by more reliable computational methods, such as MR computational methods [21,22,23,24,25,26,27,28,29,30]. However, conventional MR computational methods are intractable for large electronic systems (e.g., C-Belt[n] with large values of n), limiting the applications of these MR methods to relatively small electronic systems.
Aiming to unlock the ground-state properties of large-scale MR electronic systems, TAO-DFT (thermally-assisted-occupation density functional theory) [31] has been formulated in recent years. TAO-DFT can offer approximate static correlation in a cost-effective way, i.e., via an entropy contribution consisting of the TAO (thermally-assisted-occupation) orbital occupation numbers produced by the Fermi–Dirac distribution with a fictitious temperature , even when adopting the unsophisticated LDA (local density approximation) exchange-correlation and -dependent energy functionals [31], wherein the energy density depends only on the local electron density. More complicated semilocal and hybrid functionals in TAO-DFT have been proposed as well [32,33,34]. Moreover, the simple system-independent scheme [31,32,33] and the more sophisticated system-dependent scheme [35] in TAO-DFT have also been devised for general applications. Recently, TAO-DFT has been combined with AIMD (ab initio molecular dynamics) to further unlock the dynamical properties of large-scale MR electronic systems [36]. Because of these recent developments, TAO-DFT has been successfully used to study several challenging radical molecules and nanomaterials, giving rise to the electronic properties [7,8,9,10,17,18,37,38,39,40,41], properties related to hydrogen storage [42,43,44], and vibrational frequencies [36,45].
To avoid any possible confusion with finite-temperature DFT (FT-DFT) (first proposed by Mermin [46] and powered by the Mermin–Kohn–Sham Equation [16]), we briefly describe the difference between TAO-DFT and FT-DFT. As emphasized in Ref. [31], TAO-DFT and FT-DFT may look similar, but they differ greatly in physical meanings. TAO-DFT is an electronic structure method for studying the ground-state properties of an electronic system at zero electronic temperature, while FT-DFT is developed for the thermodynamic properties of an electronic system in thermal equilibrium at a real electronic temperature . Furthermore, in TAO-DFT, the real electronic temperature is absolute zero (i.e., = 0), and the fictitious temperature , which is related to the strength of static correlation associated with the electronic system at absolute zero, can be nonvanishing, while in FT-DFT, the real electronic temperature and the fictitious temperature are the same (i.e., they are both equal to ). In other words, for the ground-state properties of electronic systems at zero electronic temperature (i.e., = 0), FT-DFT is the same as KS-DFT, while TAO-DFT (with a nonvanishing fictitious temperature ) can differ from KS-DFT. Note also that TAO-DFT (with a vanishing fictitious temperature, i.e., = 0) reduces to KS-DFT.
As a consequence, we use TAO-DFT to compute the electronic properties of C-Belt[n] (n = 4–24), reporting the singlet–triplet gaps, fundamental gaps and the associated vertical ionization potentials and electron affinities, symmetrized von Neumann entropy, occupation numbers of active TAO-orbitals, and visualization of active TAO-orbitals in this study.
2. Computational Details
3. Results and Discussion
3.1. Singlet-Triplet Gap
The nature of the ground state of a molecule (e.g., whether the molecule has a singlet or triplet ground state) can be understood from the singlet-triplet (ST) gap. Knowledge of the ST gap is also essential for many chemical processes [48,49]. Several research efforts have been made to get this quantity accurately. Experimentally, the ST gap can be obtained from phosphorescence measurements under cryogenic conditions [50,51,52]. However, for a large molecule with pronounced MR character, it remains difficult to measure the ST gap properly. Reliable theoretical calculations on the ST gap can be necessary to fill the gap. The calculation of ST gap is also demanding in understanding the singlet fission phenomenon, which can guide and motivate experimentalists.
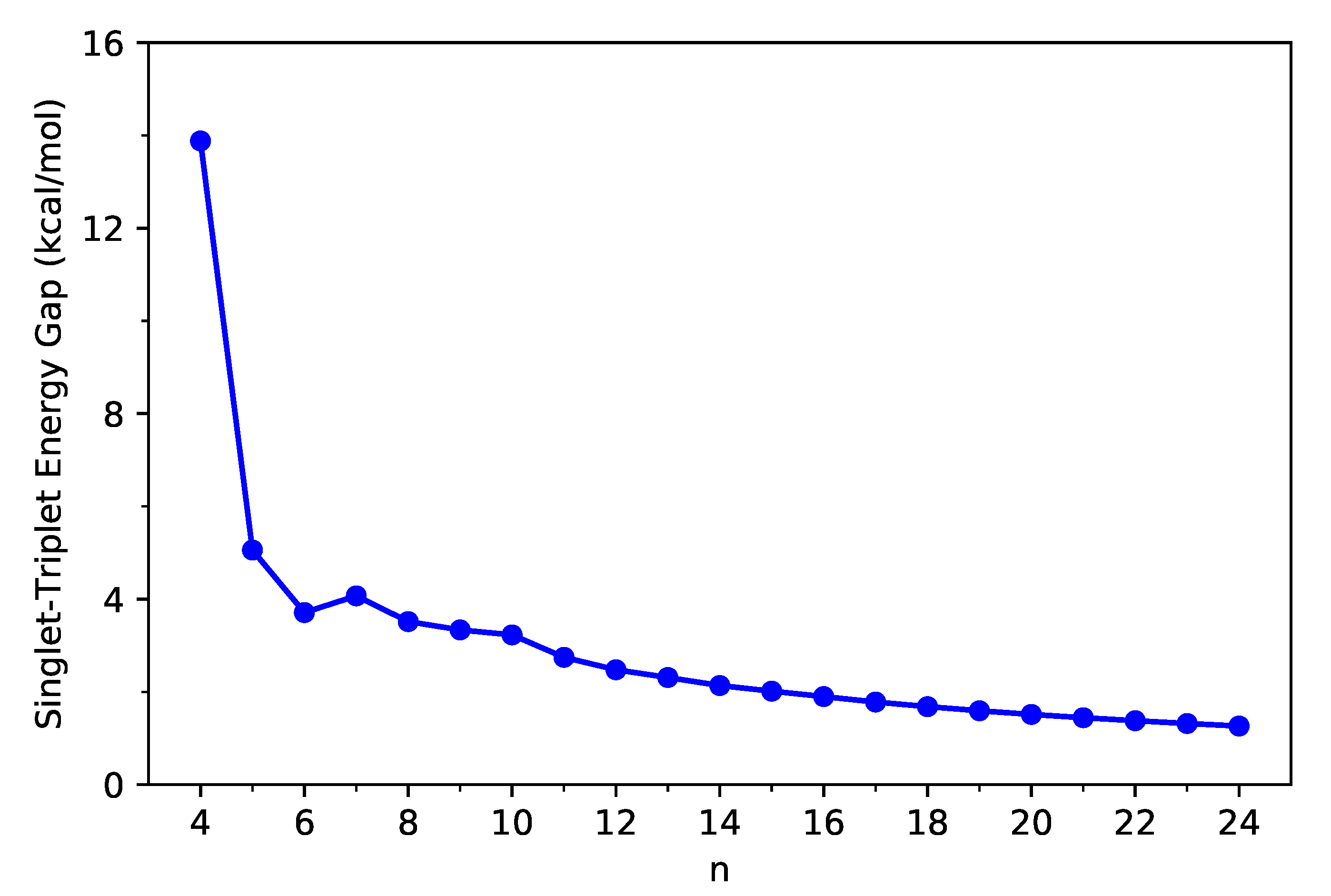
First, we calculate the ST gap () of C-Belt[n] as the difference between the lowest triplet energy and the lowest singlet energy of C-Belt[n], using spin-unrestricted TAO-LDA, where the and are evaluated on the respective optimized geometries.
As shown in Figure 2, the ground states of all the C-Belt[n] reported are singlet states (refer to Table S1 in Supplementary Information (SI) as well). The ST gap decreases drastically from n = 4 to n = 5, and for , the ST gap decreases, in a monotonic manner, with the system size n. Since MR electronic systems are often characterized by small ST gaps, the larger C-Belt[n] (e.g., ), which have very small ST gaps (e.g., less than 6 kcal/mol), are likely to possess MR character.
Here, we assess a well-known exact symmetry requirement related to MR electronic systems. For the singlet state of a MR electronic system, unphysical symmetry-breaking solutions obtained from spin-unrestricted KS-DFT with the commonly used exchange-correlation energy functionals are frequently found [19,20,31,53]. In fact, the spin-restricted and spin-unrestricted calculations using an exact theory should yield the same energy for the singlet state of the MR electronic system. In order to check whether spin-unrestricted TAO-LDA yields any unphysical symmetry-breaking solutions, we also calculate the spin-restricted TAO-LDA energies, which are evaluated on the corresponding optimized geometries, for the lowest singlet states of C-Belt[n]. The aforementioned exact symmetry requirement is found to be indeed satisfied by TAO-LDA (i.e., the spin-restricted and spin-unrestricted energies, computed using TAO-LDA, are the same within the numerical precision considered) for all the C-Belt[n] reported.
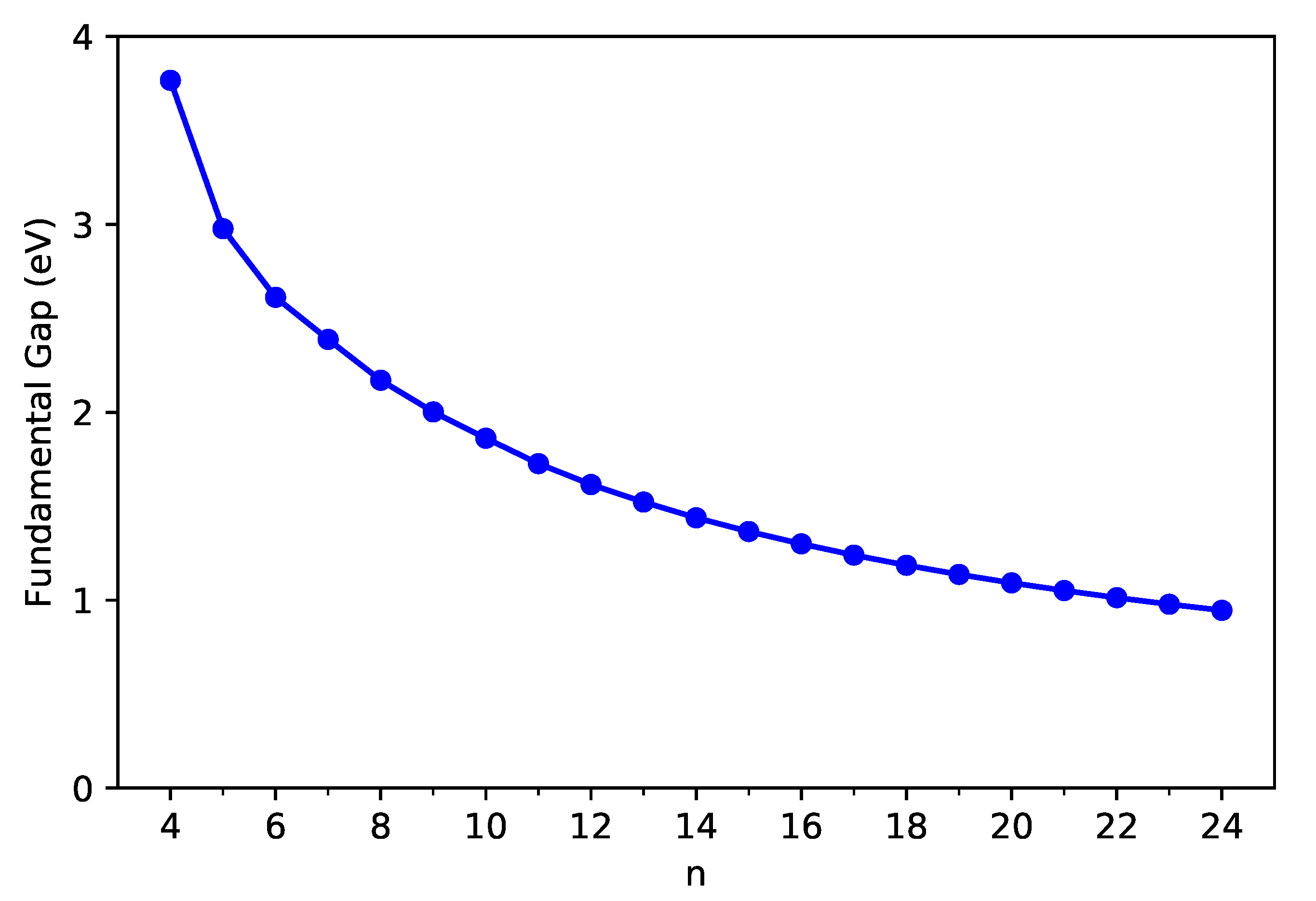
3.2. Fundamental Gap and the Associated Vertical Ionization Potential and Electron Affinity
Ionization potential (also called ionization energy) and electron affinity play an important role in several chemical processes. For a neutral molecule, the vertical ionization potential refers to the change in energy when an electron is removed from the molecule (without altering the geometry of neutral molecule), the electron affinity refers to the change in energy when an electron is added to the molecule (without altering the geometry of neutral molecule), and their difference leads to the fundamental gap.
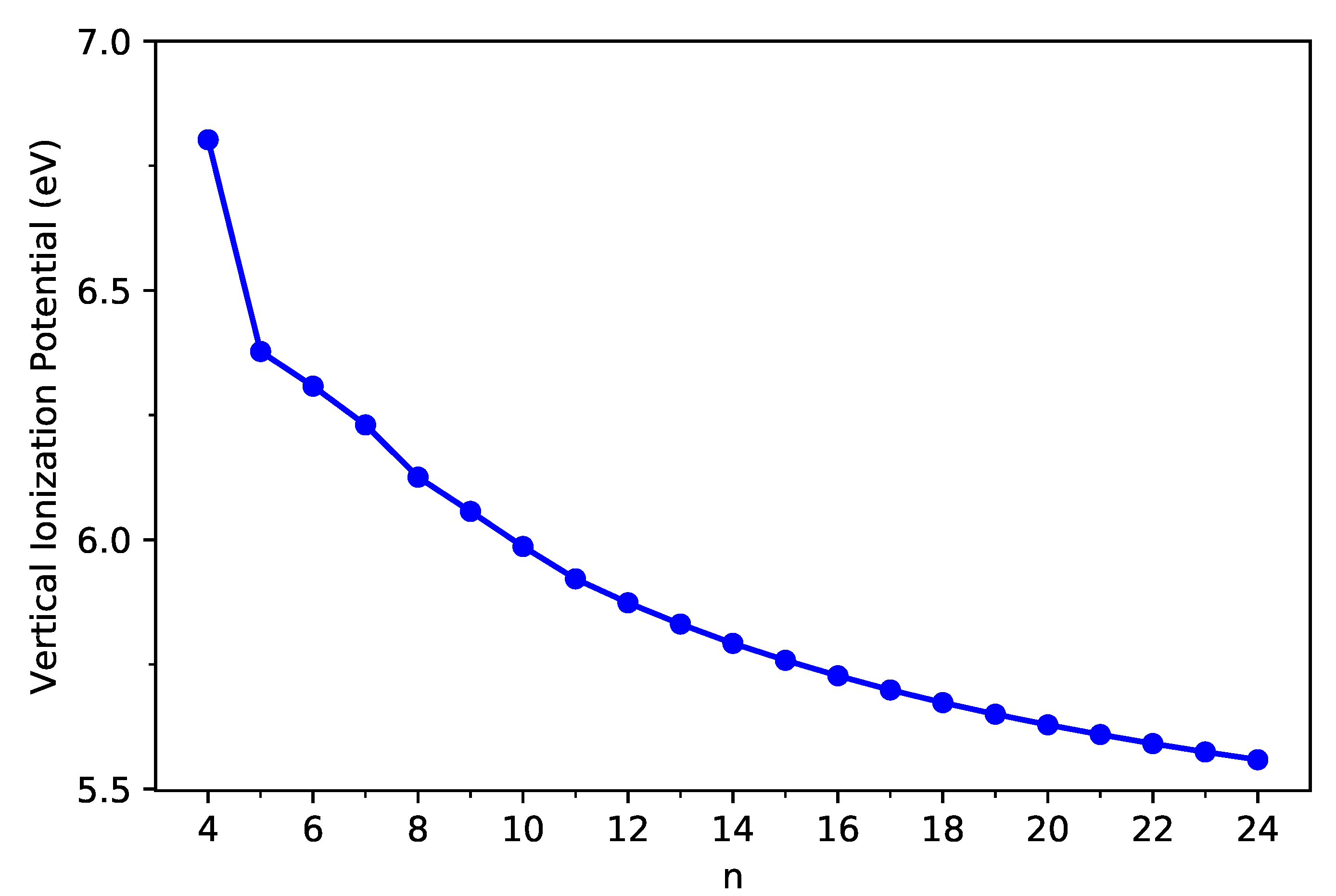
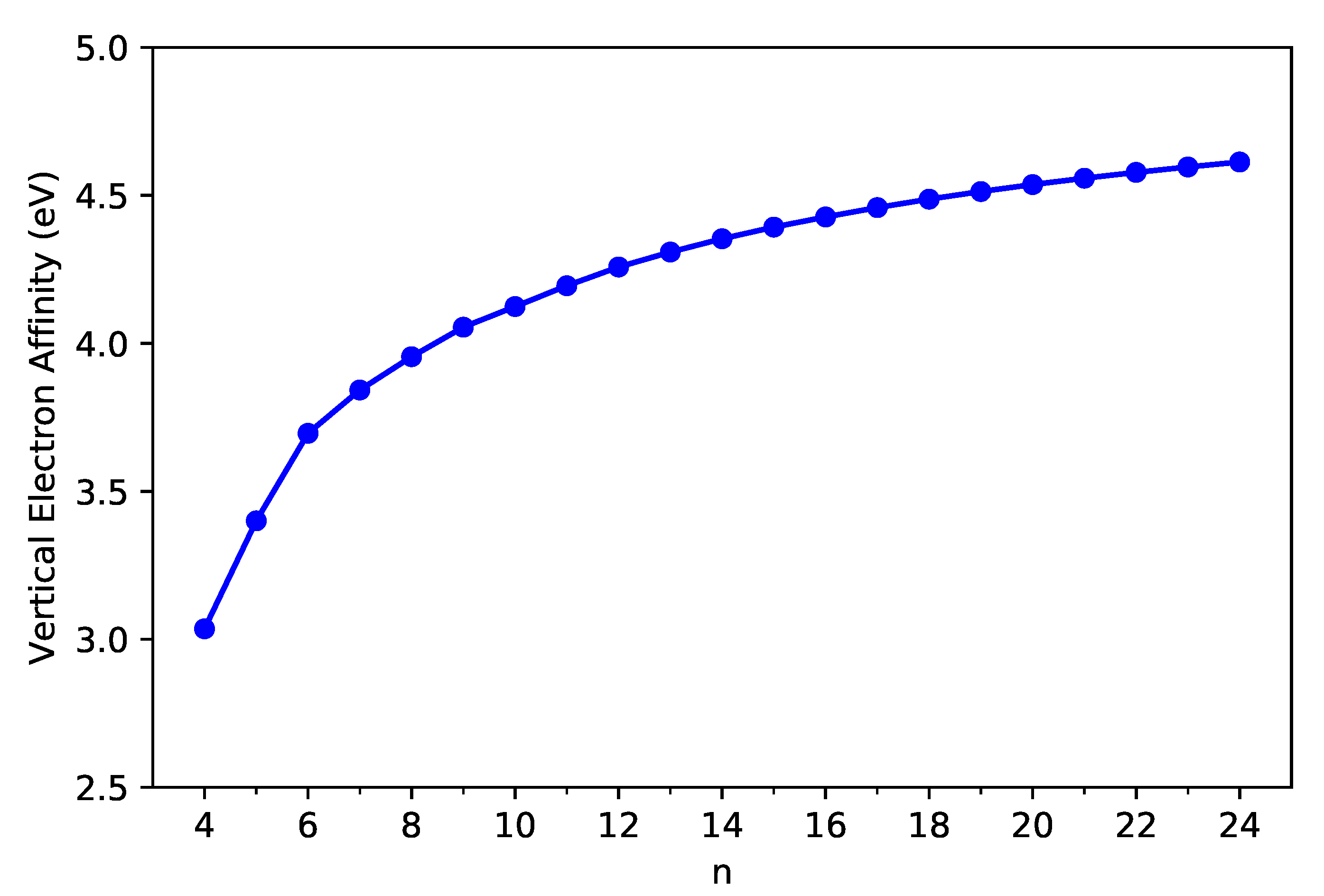
Following these definitions, with multiple energy-difference spin-unrestricted TAO-LDA calculations on the ground-state geometry of C-Belt[n], we obtain the vertical ionization potential (), the vertical electron affinity (), and the fundamental gap () of C-Belt[n], where , , and are the energies of the cationic, neutral, and anionic states, respectively, of C-Belt[n].
As shown in Figure 3 and Figure 4, the decreases monotonically, and the increases monotonically with n for all the C-Belt[n] reported. According to the definitions above, the fact that the monotonic decrease in and the monotonic increase in with the size of C-Belt[n] implies that the (see Figure 5) should decrease in a monotonic manner with n (refer to Table S2 in SI as well). The fundamental gaps of C-Belt[n] with n = 5–22 cover the range of ideal energy gaps (1–3 eV) for optoelectronic devices, and hence these carbon nanobelts can be considered as potential candidates for nanophotonics applications.
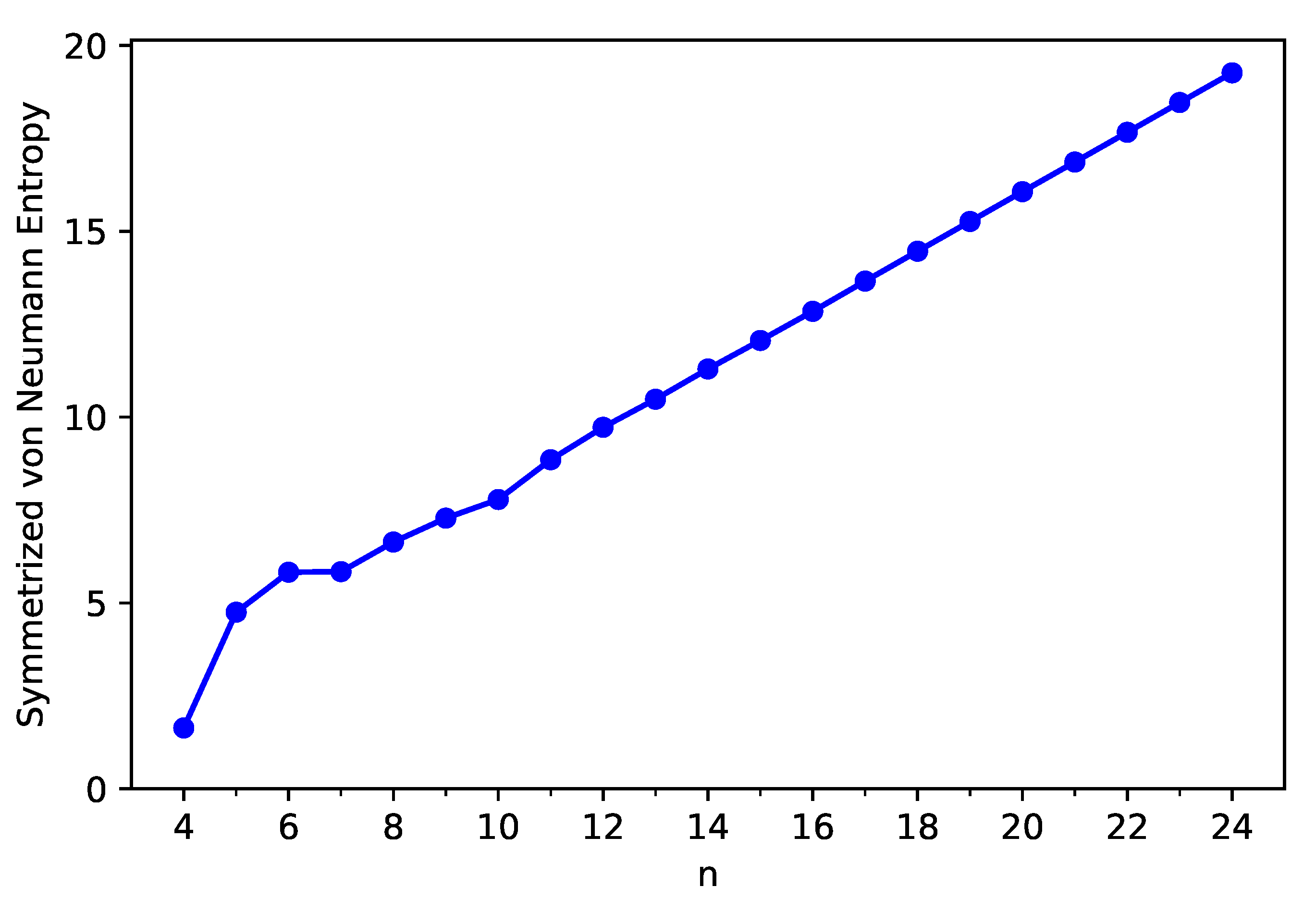
3.3. Symmetrized Von Neumann Entropy
The symmetrized von Neumann entropy [53] (see, e.g., Equation (6) of Ref. [18]),
which naturally emerges in the formulation of TAO-DFT (cf., the entropy contribution in TAO-DFT) [31,32,33], is an important quantity to understand the strength of MR character associated with an electronic system. In Equation (1), the occupation number (ranging from 0 to 1) of the -spin (e.g., up-spin or down-spin) TAO-orbital, computed using spin-unrestricted TAO-DFT [31,32,33], approximately yields the occupation number of the -spin natural orbital [54]. According to its definition, the symmetrized von Neumann entropy is vanishingly small for an electronic system with non-radical character, and can be very large for an electronic system with pronounced MR character.
To assess the strength of MR character associated with these carbon nanobelts, spin-unrestricted TAO-LDA calculations are performed to get the symmetrized von Neumann entropy of ground-state C-Belt[n]. As the size of C-Belt[n] increases, the (see Figure 6) increases in a monotonic manner (refer to Table S2 in SI as well). Moreover, the value is small for n = 4, suggesting that C-Belt[4] can be a non-radical molecule. By contrast, the values are rather large (e.g., larger than 4) for n = 5–24, suggesting that the larger C-Belt[n] (e.g., ) can exhibit MR character whose strength generally increases with n.
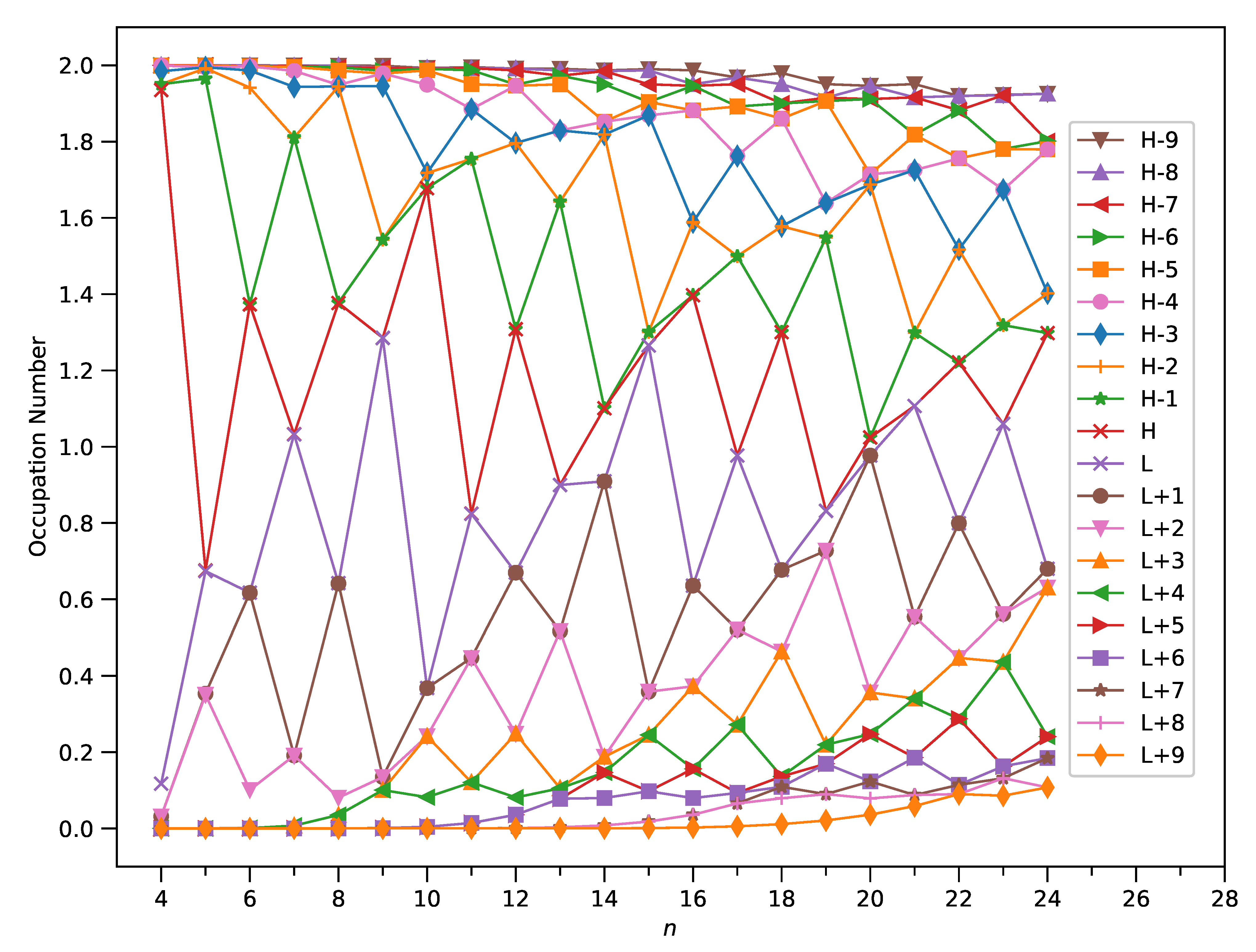
3.4. Occupation Numbers of Active TAO-Orbitals
To understand the origin of the increase of with increasing size of C-Belt[n], we plot the occupation numbers of active TAO-orbitals, such as the HOMO−9, HOMO−8, …, HOMO, LUMO, …, LUMO+8, and LUMO+9, of ground-state C-Belt[n], computed using spin-restricted TAO-LDA. For the ground state (i.e., the lowest singlet state) of C-Belt[n] (with N electrons), the TAO-orbital is taken as the HOMO, and the TAO-orbital is taken as the LUMO, and so forth. For brevity, in this work, we denote HOMO as H, LUMO as L, and so on. Note that the TAO-orbital occupation numbers (TOONs) in TAO-DFT [31,32,33] can be regarded as the approximate natural orbital occupation numbers (NOONs) [54].
As presented in Figure 7, the TOONs of ground-state C-Belt[n] clearly reveal the MR character associated with these carbon nanobelts. For example, C-Belt[4], with all the TOONs being either close to 0 or 2, is a non-radical molecule, showing consistency with the small value. Furthermore, C-Belt[7], with two TOONs being close to 1 and the remaining TOONs being either close to 0 or 2, exhibits diradical nature. In general, as n increases, C-Belt[n] possesses more TOONs deviating greatly from 0 and 2, giving rise to a more pronounced MR character and hence a larger value of .
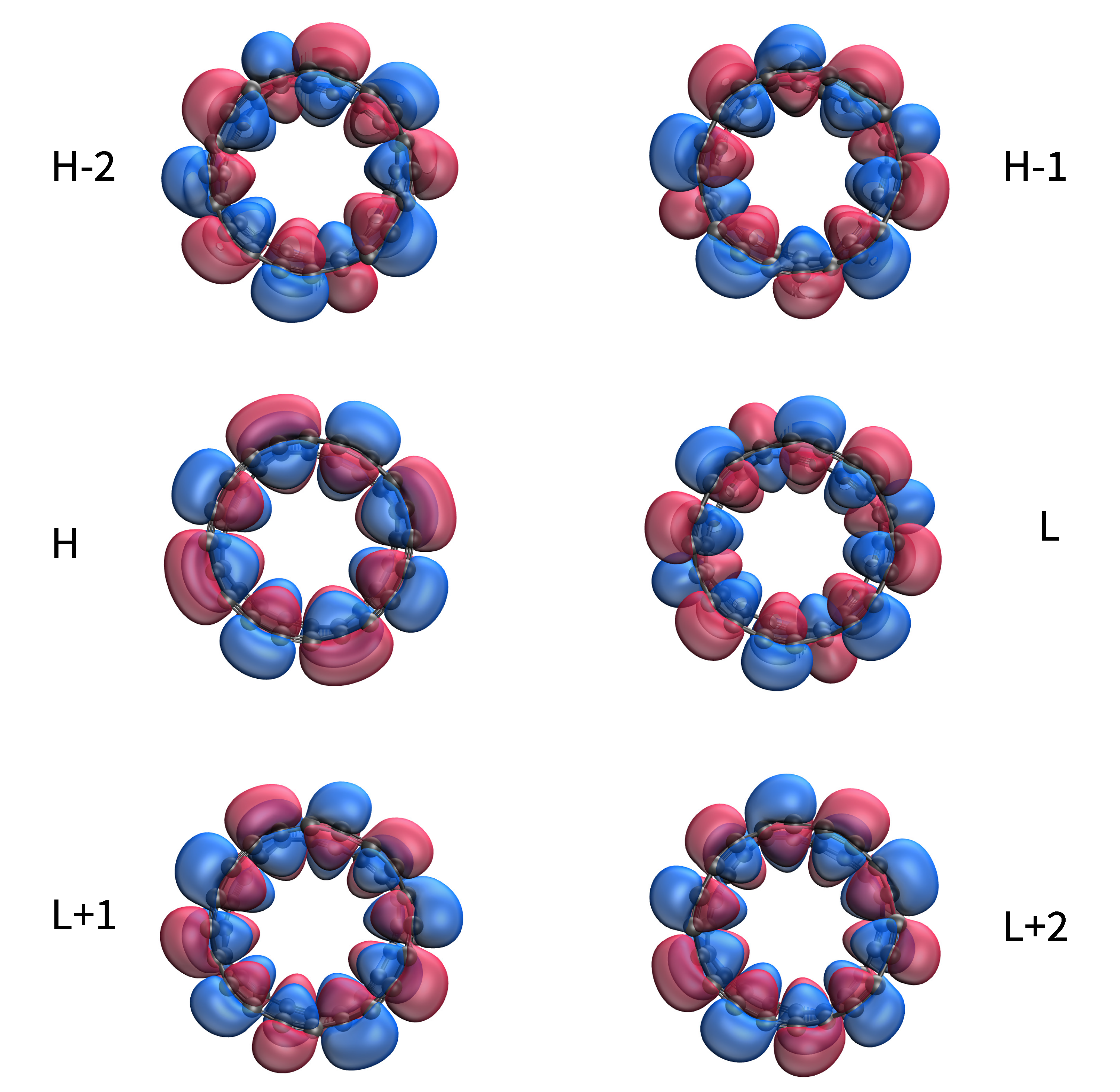
3.5. Visualization of Active TAO-Orbitals
In Figure 8, we present the visualization of active TAO-orbitals (H−2, H−1, H, L, L + 1, and L + 2) of ground-state C-Belt[4], computed using spin-restricted TAO-LDA (also refer to Figures S1–S3 in SI for the visualization of active TAO-orbitals of ground-state C-Belt[n] (n = 8, 12, and 16)). As shown, the active TAO-orbitals are delocalized over the circumference of C-Belt[n], showing similar features as have been observed in other belt-shaped molecules, such as the cyclacenes [7], cyclic boron nanoribbons [17], and cyclic carbon chains [18]. The presence of delocalized active TAO-orbitals over the C-Belt[n] implies that these carbon nanobelts can have high electrical conductivity [55,56].
4. Conclusions
In conclusion, we have presented a detailed computational study on the electronic properties of C-Belt[n] for n = 4–24. By using TAO-DFT [31], we have obtained the ST gaps, fundamental gaps and the associated vertical ionization potentials and electron affinities, symmetrized von Neumann entropy, occupation numbers of active TAO-orbitals, and visualization of active TAO-orbitals of C-Belt[n].
According to the symmetrized von Neumann entropy and the occupation numbers of active TAO-orbitals, in general, the larger C-Belt[n] should have more pronounced MR character, playing an important role in determining their electronic properties. Because of the MR character associated with these carbon nanobelts, KS-DFT with commonly used exchange-correlation energy functionals would be insufficient to predict the properties of the larger C-Belt[n] (e.g., ) in a reliable manner. Due to the computational complexity, conventional MR computational methods would be intractable, given the large size of C-Belt[n]. Relative to conventional KS-DFT and MR computational methods, TAO-DFT is especially crucial for the study of large-scale MR electronic systems, such as the series of C-Belt[n] studied here.
Because of the MR character, it can be difficult to synthesize C-Belt[n] using conventional synthesis methods. However, owing to the recent growth in the synthesis methods of belt-shaped molecules, it can be anticipated that C-Belt[n] could be realized in the near future. As a follow-up to this work, it would be interesting to further study the energetics and thermal stability of C-Belt[n] using TAO-DFT-based AIMD simulations [36], and assess these properties for potential applications. Moreover, in this work, we have reported only the electronic properties of C-Belt[n], computed using the commonly used TAO-LDA (with = 7 mhartree) [31]. Certainly, it will be interesting to further examine how these electronic properties and other properties (e.g., structural properties) vary with different values (e.g., = 0 for KS-DFT, other system-independent values [31,32,33] as well as system-dependent values [35]) and density functionals (e.g., semilocal [32] and hybrid [33] functionals) in TAO-DFT. It is worth mentioning that other efficient electronic structure methods for strong static correlation [57,58] may also be employed here. In addition to C-Belt[n] (i.e., the carbon nanobelts consisting of n fused 12-membered carbon rings) reported in this work, the electronic properties of closely related carbon allotropes, such as the carbon nanobelts consisting of n fused 8-, 10-, and 14-membered carbon rings, can also be promising for certain applications. We plan to pursue some of these directions in the near future.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10
.3390/nano11092224/s1, Figure S1: Visualization of active TAO-orbitals, such as H−2 (1.945), H−1 (1.377), H (1.377), L (0.642), L+1 (0.641), and L+2 (0.081), of ground-state C-Belt[8] at an isovalue of 0.02 e/Å, computed using spin-restricted TAO-LDA. Numbers in parentheses show the TOONs, Figure S2: Visualization of active TAO-orbitals, such as H−2 (1.796), H−1 (1.308), H (1.308), L (0.670), L+1 (0.670), and L+2 (0.249), of ground-state C-Belt[12] at an isovalue of 0.02 e/Å, computed using spin-restricted TAO-LDA. Numbers in parentheses show the TOONs, Figure S3: Visualization of active TAO-orbitals, such as H−2 (1.588), H−1 (1.397), H (1.397), L (0.636), L+1 (0.636), and L+2 (0.373), of ground-state C-Belt[16] at an isovalue of 0.02 e/Å, computed using spin-restricted TAO-LDA. Numbers in parentheses show the TOONs, Table S1: Singlet-triplet gap (in kcal/mol) of C-Belt[n], computed using spin-unrestricted TAO-LDA, Table S2: Vertical ionization potential (in eV), vertical electron affinity (in eV), fundamental gap (in eV), and symmetrized von Neumann entropy of ground-state C-Belt[n], computed using spin-unrestricted TAO-LDA.
Author Contributions
Conceptualization, S.S. and J.-D.C.; Data curation, S.S.; Formal analysis, S.S.; Funding acquisition, J.-D.C.; Investigation, S.S.; Methodology, S.S. and J.-D.C.; Project administration, J.-D.C.; Resources, J.-D.C.; Software, J.-D.C.; Supervision, J.-D.C.; Validation, S.S. and J.-D.C.; Visualization, S.S.; Writing—original draft, S.S.; Writing—review and editing, J.-D.C. Both authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Technology of Taiwan (Grant Nos. MOST110-2112-M-002-045-MY3; MOST107-2628-M-002-005-MY3).
Acknowledgments
This work was supported by the Ministry of Science and Technology of Taiwan (Grant Nos. MOST110-2112-M-002-045-MY3; MOST107-2628-M-002-005-MY3), National Taiwan University, and the National Center for Theoretical Sciences of Taiwan.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Georgakilas, V.; Perman, J.A.; Tucek, J.; Zboril, R. Broad family of carbon nanoallotropes: Classification, chemistry, and applications of fullerenes, carbon dots, nanotubes, graphene, nanodiamonds, and combined superstructures. Chem. Rev. 2015, 115, 4744–4822. [Google Scholar] [CrossRef] [PubMed]
- Taharam, K.; Tobe, Y. Molecular loops and belts. Chem. Rev. 2006, 106, 5274–5290. [Google Scholar] [CrossRef] [PubMed]
- Jasti, R.; Bhattacharjee, J.; Neaton, J.B.; Bertozzi, C.R. Synthesis, characterization, and theory of [9]−, [12]−, and [18]Cycloparaphenylene: Carbon nanohoop structures. J. Am. Chem. Soc. 2008, 130, 17646–17647. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, T.; Watanabe, Y.; Sakamoto, Y.; Suzuki, T.; Yamago, S. Selective and random syntheses of [n]Cycloparaphenylenes (n=8–13) and size dependence of their electronic properties. J. Am. Chem. Soc. 2011, 133, 8354–8361. [Google Scholar] [CrossRef]
- Estrada, E.; Simón-Manso, Y. Escherynes: Novel carbon allotropes with belt shapes. Chem. Phys. Lett. 2012, 548, 80–84. [Google Scholar] [CrossRef]
- Wu, C.-S.; Lee, P.-Y.; Chai, J.-D. Electronic properties of cyclacenes from TAO-DFT. Sci. Rep. 2016, 6, 37249. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.-H.; Chai, J.-D. Electronic properties of Möbius cyclacenes studied by thermally-assisted-occupation density functional theory. Sci. Rep. 2019, 9, 2907. [Google Scholar] [CrossRef] [Green Version]
- Hanson-Heine, M.W.D.; Rogers, D.M.; Woodward, S.; Hirst, J.D. Dewar benzenoids discovered in carbon nanobelts. J. Phys. Chem. Lett. 2020, 11, 3769–3772. [Google Scholar] [CrossRef]
- Hanson-Heine, M.W.D.; Hirst, J.D. Möbius and Hückel cyclacenes with Dewar and Ladenburg defects. J. Phys. Chem. A 2020, 124, 5408–5414. [Google Scholar] [CrossRef]
- Cheung, K.Y.; Watanabe, K.; Segawa, Y.; Itami, K. Synthesis of a zigzag carbon nanobelt. Nat. Chem. 2021, 13, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Four- and eight-membered rings carbon nanotubes: A new class of carbon nanomaterials. Results Phys. 2018, 9, 656–664. [Google Scholar] [CrossRef]
- Bunz, U.H.; Rubin, Y.; Tobe, Y. Polyethynylated cyclic π-systems: Scaffoldings for novel two and three-dimensional carbon networks. Chem. Soc. Rev. 1999, 28, 107–119. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Y.E.; Tong, S.; Wang, M.X. Hydrocarbon belts with truncated cone structures. J. Am. Chem. Soc. 2020, 142, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Povie, G.; Segawa, Y.; Nishihara, T.; Miyauchi, Y.; Itami, K. Synthesis of a carbon nanobelt. Science 2017, 356, 172–175. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Seenithurai, S.; Chai, J.-D. Electronic properties of linear and cyclic boron nanoribbons from thermally-assisted-occupation density functional theory. Sci. Rep. 2019, 9, 12139. [Google Scholar] [CrossRef] [PubMed]
- Seenithurai, S.; Chai, J.-D. TAO-DFT investigation of electronic properties of linear and cyclic carbon chains. Sci. Rep. 2020, 10, 13133. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into current limitations of density functional theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Challenges for density functional theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.-Å.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218. [Google Scholar] [CrossRef]
- Hachmann, J.; Dorando, J.J.; Aviles, M.; Chan, G.K.L. The radical character of the acenes: A density matrix renormalization group study. J. Chem. Phys. 2007, 127, 134309. [Google Scholar] [CrossRef] [Green Version]
- Gidofalvi, G.; Mazziotti, D.A. Active-space two-electron reduced-density-matrix method: Complete active-space calculations without diagonalization of the N-electron hamiltonian. J. Chem. Phys. 2008, 129, 134108. [Google Scholar] [CrossRef]
- Mizukami, W.; Kurashige, Y.; Yanai, T. More π electrons make a difference: Emergence of many radicals on graphene nanoribbons studied by ab initio DMRG theory. J. Chem. Theory Comput. 2013, 9, 401–407. [Google Scholar] [CrossRef]
- Gryn’ova, G.; Coote, M.L.; Corminboeuf, C. Theory and practice of uncommon molecular electronic configurations. WIREs Comput. Mol. Sci. 2015, 5, 440–459. [Google Scholar] [CrossRef] [Green Version]
- Fosso-Tande, J.; Nguyen, T.-S.; Gidofalvi, G.; DePrince, A.E., III. Large-scale variational two-electron reduced-density-matrix-driven complete active space self-consistent field methods. J. Chem. Theory Comput. 2016, 12, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Piris, M. Global method for electron correlation. Phys. Rev. Lett. 2017, 119, 063002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meer, R.; Gritsenko, O.V.; Baerends, E.J. A non-JKL density matrix functional for intergeminal correlation between closed-shell geminals from analysis of natural orbital configuration interaction expansions. J. Chem. Phys. 2018, 148, 104102. [Google Scholar] [CrossRef]
- Van Meer, R.; Chai, J.-D. Approximate density matrix functionals applied to hetero-atomic bond dissociation. Eur. Phys. J. B 2020, 93, 172. [Google Scholar] [CrossRef]
- Van Meer, R.; Gritsenko, O.; Chai, J.-D. Combining density-based dynamical correlation with a reduced-density-matrix strong-correlation description. Phys. Rev. A 2020, 102, 032815. [Google Scholar] [CrossRef]
- Chai, J.-D. Density functional theory with fractional orbital occupations. J. Chem. Phys. 2012, 136, 154104. [Google Scholar] [CrossRef]
- Chai, J.-D. Thermally-assisted-occupation density functional theory with generalized-gradient approximations. J. Chem. Phys. 2014, 140, 18A521. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.-D. Role of exact exchange in thermally-assisted-occupation density functional theory: A proposal of new hybrid schemes. J. Chem. Phys. 2017, 146, 044102. [Google Scholar] [CrossRef] [Green Version]
- Xuan, F.; Chai, J.-D.; Su, H. Local density approximation for the short-range exchange free energy functional. ACS Omega 2019, 4, 7675–7683. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Hui, K.; Chung, J.-H.; Chai, J.-D. Self-consistent determination of the fictitious temperature in thermally-assisted-occupation density functional theory. RSC Adv. 2017, 7, 50496–50507. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Chai, J.-D. TAO-DFT-based ab initio molecular dynamics. Front. Chem. 2020, 8, 589432. [Google Scholar] [CrossRef]
- Wu, C.-S.; Chai, J.-D. Electronic properties of zigzag graphene nanoribbons studied by TAO-DFT. J. Chem. Theory Comput. 2015, 11, 2003–2011. [Google Scholar] [CrossRef]
- Yeh, C.-N.; Chai, J.-D. Role of Kekulé and non-Kekulé structures in the radical character of alternant polycyclic aromatic hydrocarbons: A TAO-DFT study. Sci. Rep. 2016, 6, 30562. [Google Scholar] [CrossRef]
- Yeh, C.-N.; Wu, C.; Su, H.; Chai, J.-D. Electronic properties of the coronene series from thermally-assisted-occupation density functional theory. RSC Adv. 2018, 8, 34350. [Google Scholar] [CrossRef] [Green Version]
- Deng, Q.; Chai, J.-D. Electronic properties of triangle-shaped graphene nanoflakes from TAO-DFT. ACS Omega 2019, 4, 14202–14210. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-J.; Seenithurai, S.; Chai, J.-D. TAO-DFT study on the electronic properties of diamond-shaped graphene nanoflakes. Nanomaterials 2020, 10, 1236. [Google Scholar] [CrossRef]
- Seenithurai, S.; Chai, J.-D. Effect of Li adsorption on the electronic and hydrogen storage properties of acenes: A dispersion-corrected TAO-DFT study. Sci. Rep. 2016, 6, 33081. [Google Scholar] [CrossRef] [Green Version]
- Seenithurai, S.; Chai, J.-D. Effect of Li termination on the electronic and hydrogen storage properties of linear carbon chains: A TAO-DFT study. Sci. Rep. 2017, 7, 4966. [Google Scholar] [CrossRef] [Green Version]
- Seenithurai, S.; Chai, J.-D. Electronic and hydrogen storage properties of Li-terminated linear boron chains studied by TAO-DFT. Sci. Rep. 2018, 8, 13538. [Google Scholar] [CrossRef]
- Hanson-Heine, M.W.D. Static correlation in vibrational frequencies studied using thermally-assisted-occupation density functional theory. Chem. Phys. Lett. 2020, 739, 137012. [Google Scholar] [CrossRef]
- Mermin, N.D. Thermal properties of the inhomogeneous electron gas. Phys. Rev. 1965, 137, A1441. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Wang, X.; Wang, L.; Zhang, Z.; Wang, X.; Song, Y.; Power, P.P. Thermally controlling the singlet-triplet energy gap of a diradical in the solid state. Chem. Sci. 2016, 7, 6514–6518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Wang, X.; Wang, L.; Zhang, Z.; Wang, X.; Song, Y.; Power, P.P. Achieving a balance between small singlet-triplet energy splitting and high fluorescence radiative rate in a quinoxaline-based orange-red thermally activated delayed fluorescence emitter. Chem. Commun. 2016, 52, 11012–11015. [Google Scholar]
- Smith, M.B.; Michl, J. Singlet fission. Chem. Rev. 2010, 110, 6891–6936. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, Q.; Feng, W.; Sun, Y.; Li, F. Upconversion luminescent materials: Advances and applications. Chem. Rev. 2015, 115, 395–465. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic photoredox catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Rivero, P.; Jiménez-Hoyos, C.A.; Scuseria, G.E. Entanglement and polyradical nature of polycyclic aromatic hydrocarbons predicted by projected Hartree-Fock theory. J. Phys. Chem. B 2013, 117, 12750–12758. [Google Scholar] [CrossRef]
- Löwdin, P.-O.; Shull, H. Natural orbitals in the quantum theory of two-electron systems. Phys. Rev. 1956, 101, 1730–1739. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure; Wiley: New York, NY, USA, 2007; pp. 3–380. [Google Scholar]
- Che, Y.; Datar, A.; Yang, X.; Naddo, T.; Zhao, J.; Zang, L. Enhancing one-dimensional charge transport through intermolecular π-electron delocalization: Conductivity improvement for organic nanobelts. J. Am. Chem. Soc. 2007, 129, 6354–6355. [Google Scholar] [CrossRef]
- Lathiotakis, N.N.; Helbig, N.; Rubio, A.; Gidopoulos, N.I. Local reduced-density-matrix-functional theory: Incorporating static correlation effects in Kohn-Sham equations. Phys. Rev. A 2014, 90, 032511. [Google Scholar] [CrossRef] [Green Version]
- Theophilou, I.; Lathiotakis, N.N.; Gidopoulos, N.I.; Rubio, A.; Helbig, N. Orbitals from local RDMFT: Are they Kohn-Sham or natural orbitals? J. Chem. Phys. 2015, 143, 054106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
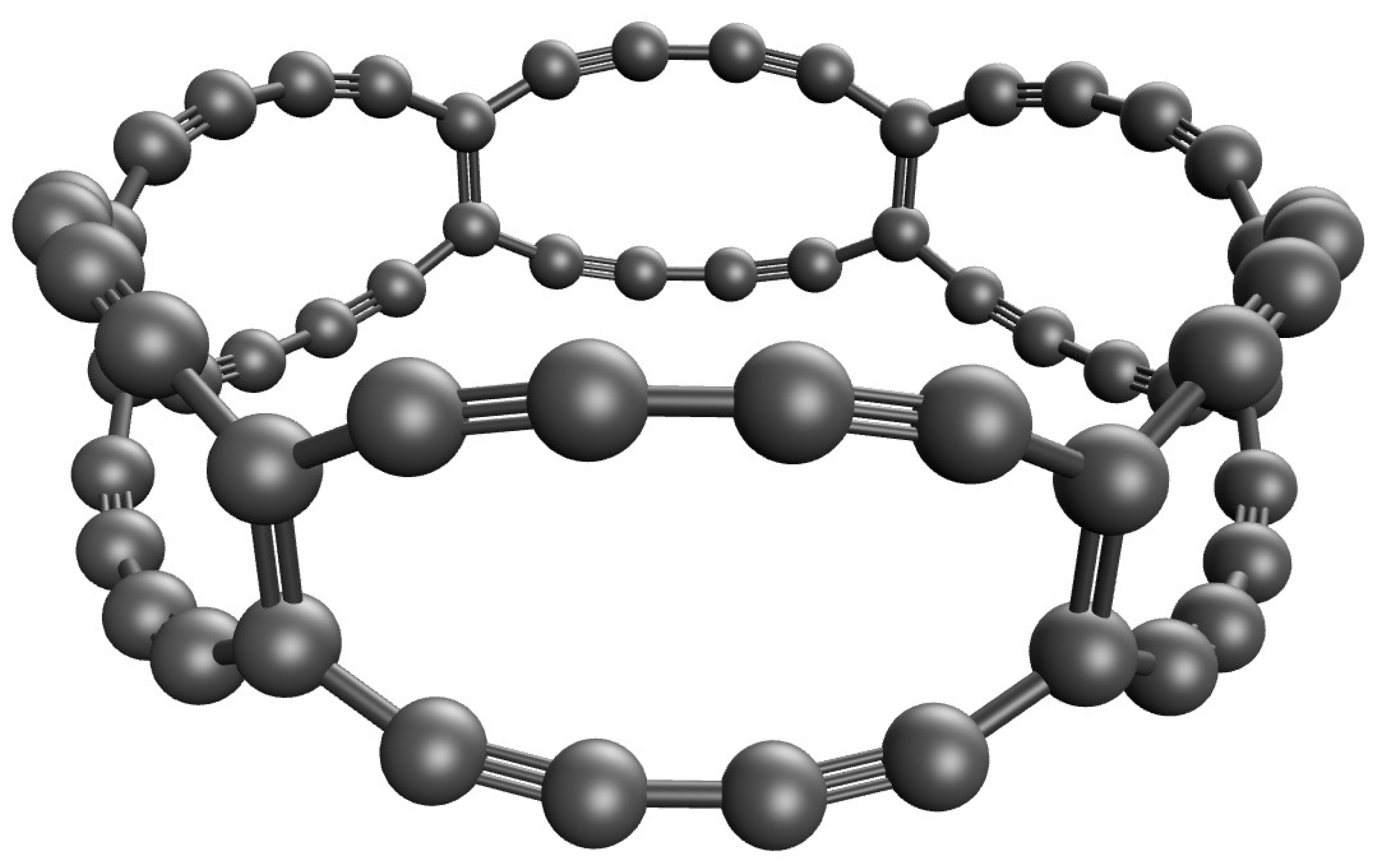
Figure 1.
Geometry of C-Belt[6], containing 6 fused 12-membered carbon rings forming a closed loop.

Figure 2.
Singlet–triplet gap of C-Belt[n], computed using spin-unrestricted TAO-LDA.

Figure 3.
Vertical ionization potential of ground-state C-Belt[n], computed using spin-unrestricted TAO-LDA.
Figure 3.
Vertical ionization potential of ground-state C-Belt[n], computed using spin-unrestricted TAO-LDA.

Figure 4.
Vertical electron affinity of ground-state C-Belt[n], computed using spin-unrestricted TAO-LDA.
Figure 4.
Vertical electron affinity of ground-state C-Belt[n], computed using spin-unrestricted TAO-LDA.

Figure 5.
Fundamental gap of ground-state C-Belt[n], computed using spin-unrestricted TAO-LDA.

Figure 6.
Symmetrized von Neumann entropy of ground-state C-Belt[n], computed using spin-unrestricted TAO-LDA.
Figure 6.
Symmetrized von Neumann entropy of ground-state C-Belt[n], computed using spin-unrestricted TAO-LDA.

Figure 7.
Occupation numbers of active TAO-orbitals (H − 9, H − 8, …, H − 1, H, L, L+1, …, L + 8, and L + 9) of ground-state C-Belt[n], computed using spin-restricted TAO-LDA.
Figure 7.
Occupation numbers of active TAO-orbitals (H − 9, H − 8, …, H − 1, H, L, L+1, …, L + 8, and L + 9) of ground-state C-Belt[n], computed using spin-restricted TAO-LDA.

Figure 8.
Visualization of active TAO-orbitals, such as H−2 (1.951), H−1 (1.950), H (1.935), L (0.117), L + 1 (0.032), and L + 2 (0.032), of ground-state C-Belt[4] at an isovalue of 0.02 e/Å, computed using spin-restricted TAO-LDA. Numbers in parentheses show the TOONs.
Figure 8.
Visualization of active TAO-orbitals, such as H−2 (1.951), H−1 (1.950), H (1.935), L (0.117), L + 1 (0.032), and L + 2 (0.032), of ground-state C-Belt[4] at an isovalue of 0.02 e/Å, computed using spin-restricted TAO-LDA. Numbers in parentheses show the TOONs.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Seenithurai, S.; Chai, J.-D. Electronic Properties of Carbon Nanobelts Predicted by Thermally-Assisted-Occupation DFT. Nanomaterials 2021, 11, 2224. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11092224
AMA Style
Seenithurai S, Chai J-D. Electronic Properties of Carbon Nanobelts Predicted by Thermally-Assisted-Occupation DFT. Nanomaterials. 2021; 11(9):2224. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11092224
Chicago/Turabian StyleSeenithurai, Sonai, and Jeng-Da Chai. 2021. "Electronic Properties of Carbon Nanobelts Predicted by Thermally-Assisted-Occupation DFT" Nanomaterials 11, no. 9: 2224. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11092224
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.