
Free-Standing, Interwoven Tubular Graphene Mesh-Supported Binary AuPt Nanocatalysts: An Innovative and High-Performance Anode Methanol Oxidation Catalyst

 ,
,  and
and
Abstract
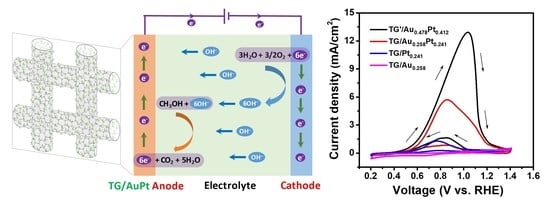
:
1. Introduction
2. Experimental Section
2.1. Preparation of the Catalyst Supporting Material: TG Meshes
2.2. Catalysts Loading on the Supporting Material
2.2.1. Loading Au NPs on TG Meshes
2.2.2. Loading Pt NPs on TG Meshes
2.2.3. Loading Binary AuPt NPs on TG Meshes
2.3. Characterization
3. Results and Discussion
3.1. Material Characterization
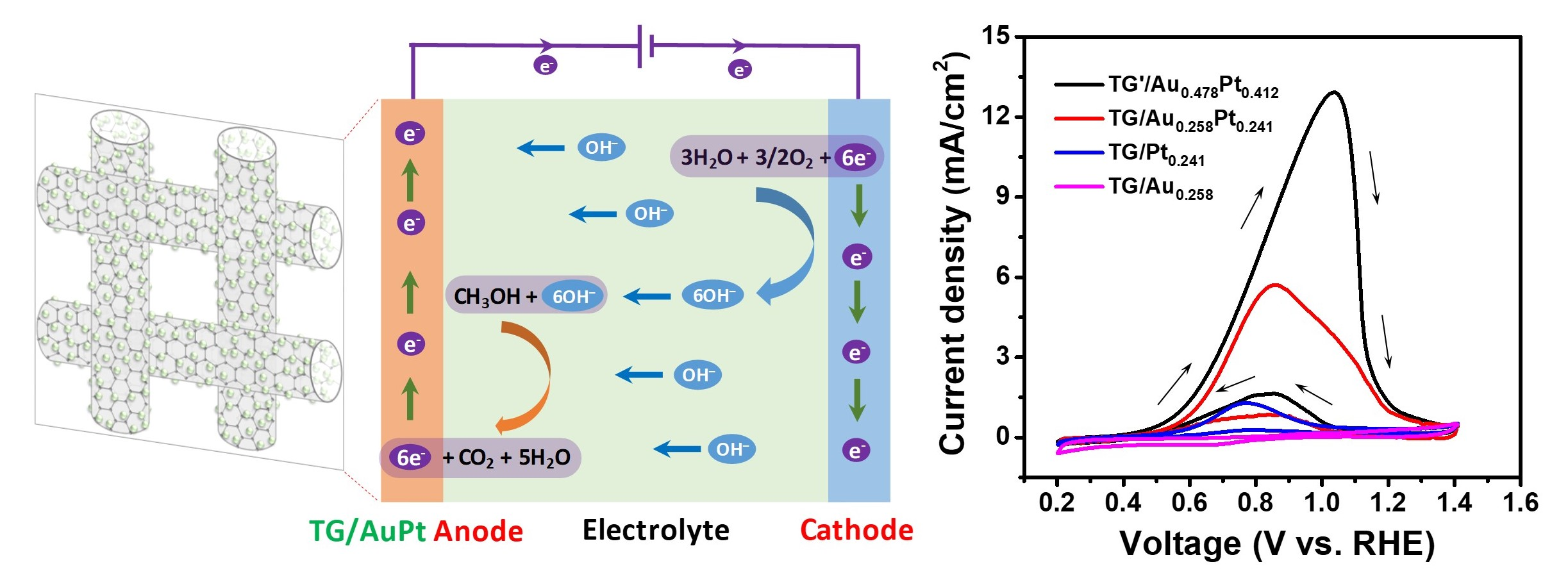
3.2. Methanol Oxidation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaur, A.; Kaur, G.; Singh, P.P.; Kaushal, S. Supported Bimetallic Nanoparticles as Anode Catalysts for Direct Methanol Fuel Cells: A Review. Int. J. Hydrog. Energy 2021, 46, 15820–15849. [Google Scholar] [CrossRef]
- Shrivastava, N.K.; Harris, T.A.L. Direct Methanol Fuel Cells. In Encyclopedia of Sustainable Technologies; Martin, A.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 343–357. [Google Scholar]
- Joghee, P.; Malik, J.N.; Pylypenko, S.; O’Hayre, R. A Review on Direct Methanol Fuel Cells—In the Perspective of Energy and Sustainability. MRS Energy Sustain. 2015, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Breeze, P. Chapter 8—Direct Methanol Fuel Cell. In Feal Cells; Breeze, P., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 75–82. [Google Scholar]
- Samimi, F.; Rahimpour, M.R. Chapter 14—Direct Methanol Fuel Cell. In Methanol: Sience and Engineering; Basile, A., Dalena, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 381–397. [Google Scholar]
- Abraham, B.G.; Chetty, R. Design and Fabrication of a Quick-Fit Architecture Air Breathing Direct Methanol Fuel Cell. Int. J. Hydrog. Energy 2021, 46, 6845–6856. [Google Scholar] [CrossRef]
- Qiu, X.; Yan, X.; Cen, K.; Sun, D.; Xu, L.; Tang, Y. Achieving Highly Electrocatalytic Performance by Constructing Holey Reduced Graphene Oxide Hollow Nanospheres Sandwiched by Interior and Exterior Platinum Nanoparticles. ACS Appl. Energy Mater. 2018, 1, 2341–2349. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Ghosh, S.; Bhattachrya, S.K. Improved Catalysis of Green-Synthesized Pd-Ag Alloy-Nanoparticles for Anodic Oxidation of Methanol in Alkali. Electrochim. Acta 2017, 225, 310–321. [Google Scholar] [CrossRef]
- Lee, D.; Gok, S.; Kim, Y.; Sung, Y.-E.; Lee, E.; Jang, J.-H.; Kwon, O.J.; Lim, T. Methanol Tolerant Pt–C Core–Shell Cathode Catalyst for Direct Methanol Fuel Cells. ACS Appl. Mater. Interfaces 2020, 12, 44588–44596. [Google Scholar] [CrossRef]
- Johánek, V.; Ostroverkh, A.; Fiala, R. Vapor-Feed Low Temperature Direct Methanol Fuel Cell with Pt and PtRu Electrodes: Chemistry Insight. Renew. Energy 2019, 138, 409–415. [Google Scholar] [CrossRef]
- Zhang, S.; Rong, H.; Yang, T.; Bai, B.; Zhang, J. Ultrafine PtRu Dilute Alloy Nanodendrites for Enhanced Electrocatalytic Methanol Oxidation. Chem.—Eur. J. 2020, 26, 4025–4031. [Google Scholar] [CrossRef]
- Bai, J.; Xiao, X.; Xue, Y.-Y.; Jiang, J.-X.; Zeng, J.-H.; Li, X.-F.; Chen, Y. Bimetallic Platinum–Rhodium Alloy Nanodendrites as Highly Active Electrocatalyst for the Ethanol Oxidation Reaction. ACS Appl. Mater. Interfaces 2018, 10, 19755–19763. [Google Scholar] [CrossRef]
- Xie, Y.; Li, C.; Razek, S.A.; Fang, J.; Dimitrov, N. Synthesis of Nanoporous Au−Cu−Pt Alloy as a Superior Catalyst for the Methanol Oxidation Reaction. ChemElectroChem 2020, 7, 569–580. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, K.; Qiu, J.; Wu, J.; Shao, J.; Wang, H.; Zhang, Y.; Han, J.; Zhang, Y.; Yan, L. Ternary PtFeCo Alloys on Graphene with High Electrocatalytic Activities for Methanol Oxidation. Nanoscale 2020, 12, 9824–9832. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Ouyang, J. Ternary NiAuPt Nanoparticles on Reduced Graphene Oxide as Catalysts toward the Electrochemical Oxidation Reaction of Ethanol. ACS Catal. 2015, 5, 1371–1380. [Google Scholar] [CrossRef]
- Fahim, A.E.; Abdel Hameed, R.M.; Allam, N.K. Synthesis and Characterization of Core–Shell Structured M@Pd/SnO2–Graphene [M = Co, Ni or Cu] Electrocatalysts for Ethanol Oxidation in Alkaline Solution. New J. Chem. 2018, 42, 6144–6160. [Google Scholar] [CrossRef]
- Vilian, A.T.E.; Hwang, S.-K.; Kwak, C.H.; Oh, S.Y.; Kim, C.-Y.; Lee, G.; Lee, J.B.; Huh, Y.S.; Han, Y.-K. Pt-Au Bimetallic Nanoparticles Decorated on Reduced Graphene Oxide as an Excellent Electrocatalysts for Methanol Oxidation. Synth. Met. 2016, 219, 52–59. [Google Scholar] [CrossRef]
- Ghosh, S.; Bera, S.; Bysakh, S.; Basu, R.N. Conducting Polymer Nanofiber-Supported Pt Alloys: Unprecedented Materials for Methanol Oxidation with Enhanced Electrocatalytic Performance and Stability. Sustain. Energy Fuels 2017, 1, 1148–1161. [Google Scholar] [CrossRef]
- Ghosh, S.; Bera, S.; Bysakh, S.; Basu, R.N. Highly Active Multimetallic Palladium Nanoalloys Embedded in Conducting Polymer as Anode Catalyst for Electrooxidation of Ethanol. ACS Appl. Mater. Interfaces 2017, 9, 33775–33790. [Google Scholar] [CrossRef]
- Zhang, H.; Ren, W.; Guan, C.; Cheng, C. Pt Decorated 3D Vertical Graphene Nanosheet Arrays for Efficient Methanol Oxidation and Hydrogen Evolution Reactions. J. Mater. Chem. A 2017, 5, 22004–22011. [Google Scholar] [CrossRef]
- Sharma, S.; Pollet, B.G. Support Materials for PEMFC and DMFC Electrocatalysts—A Review. J. Power Sources 2012, 208, 96–119. [Google Scholar] [CrossRef]
- Van Deelen, T.W.; Hernández Mejía, C.; de Jong, K.P. Control of Metal-Support Interactions in Heterogeneous Catalysts to Enhance Activity and Selectivity. Nat. Catal. 2019, 2, 955–970. [Google Scholar] [CrossRef]
- Ramli, Z.A.C.; Kamarudin, S.K. Platinum-Based Catalysts on Various Carbon Supports and Conducting Polymers for Direct Methanol Fuel Cell Applications: A Review. Nanoscale Res. Lett. 2018, 13, 410. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.-J.; Wang, A.-J.; Ma, X.; Xiang, R.-Y.; Chen, J.-R.; Feng, J.-J. One-Pot Synthesis of Porous Pt–Au Nanodendrites Supported on Reduced Graphene Oxide Nanosheets toward Catalytic Reduction of 4-Nitrophenol. J. Mater. Chem. A 2015, 3, 290–296. [Google Scholar] [CrossRef]
- Barman, B.K.; Nanda, K.K. Ultrafast-Versatile-Domestic-Microwave-Oven Based Graphene Oxide Reactor for the Synthesis of Highly Efficient Graphene Based Hybrid Electrocatalysts. ACS Sustain. Chem. Eng. 2018, 6, 4037–4045. [Google Scholar] [CrossRef]
- Bhat, S.A.; Rashid, N.; Rather, M.A.; Pandit, S.A.; Rather, G.M.; Ingole, P.P.; Bhat, M.A. PdAg Bimetallic Nanoalloy-Decorated Graphene: A Nanohybrid with Unprecedented Electrocatalytic, Catalytic, and Sensing Activities. ACS Appl. Mater. Interfaces 2018, 10, 16376–16389. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhu, J.; Li, D.; Shen, C.; Li, M.; Zhang, X.; Jiang, Q.; Zhang, J.; Wu, Y. Pt Nanoparticles Grown on 3D RuO2-Modified Graphene Architectures for Highly Efficient Methanol Oxidation. J. Mater. Chem. A 2017, 5, 4560–4567. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Lacey, S.D.; Xu, L.; Xie, H.; Li, T.; Danner, V.A.; Hu, L. Reduced Graphene Oxide Film with Record-High Conductivity and Mobility. Mater. Today 2018, 21, 186–192. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Lai, W.-C.; Tran, V.V.; Nguyen, D.D.; Kan, H.-C.; Hsu, C.-C. Tubular Graphene Architectures at the Macroscopic Scale: Fabrication and Properties. Adv. Device Mater. 2016, 2, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.D.; Suzuki, S.; Kato, S.; To, B.D.; Hsu, C.C.; Murata, H.; Rokuta, E.; Tai, N.-H.; Yoshimura, M. Macroscopic, Freestanding, and Tubular Graphene Architectures Fabricated via Thermal Annealing. ACS Nano 2015, 9, 3206–3214. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Hsieh, P.-Y.; Tsai, M.-T.; Lee, C.-Y.; Tai, N.-H.; To, B.D.; Vu, D.T.; Hsu, C.C. Hollow Few-Layer Graphene-Based Structures from Parafilm Waste for Flexible Transparent Supercapacitors and Oil Spill Cleanup. ACS Appl. Mater. Interfaces 2017, 9, 40645–40654. [Google Scholar] [CrossRef]
- Tran, V.V.; Nguyen, D.D.; Hofmann, M.; Hsieh, Y.-P.; Kan, H.-C.; Hsu, C.-C. Edge-Rich Interconnected Graphene Mesh Electrode with High Electrochemical Reactivity Applicable for Glucose Detection. Nanomaterials 2021, 11, 511. [Google Scholar] [CrossRef]
- Tran, V.V.; Nguyen, D.D.; Nguyen, A.T.; Hofmann, M.; Hsieh, Y.-P.; Kan, H.-C.; Hsu, C.-C. Electromagnetic Interference Shielding by Transparent Graphene/Nickel Mesh Films. ACS Appl. Nano Mater. 2020, 3, 7474–7481. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Lai, W.-C.; To, B.D.; Nguyen, D.D.; Hsieh, Y.-P.; Hofmann, M.; Kan, H.-C.; Hsu, C.-C. Layer Control of Tubular Graphene for Corrosion Inhibition of Nickel Wires. ACS Appl. Mater. Interfaces 2017, 9, 22911–22917. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-H.; Nguyen, A.T.; Chiu, Y.-H.; Li, J.-M.; Hsu, Y.-J. Au-Decorated GaOOH Nanorods Enhanced the Performance of Direct Methanol Fuel Cells under Light Illumination. Appl. Catal. B Environ. 2016, 185, 133–140. [Google Scholar] [CrossRef]
- Yuan, W.; Fan, X.; Cui, Z.M.; Chen, T.; Dong, Z.; Li, C.M. Controllably Self-Assembled Graphene-Supported Au@Pt Bimetallic Nanodendrites as Superior Electrocatalysts for Methanol Oxidation in Direct Methanol Fuel Cells. J. Mater. Chem. A 2016, 4, 7352–7364. [Google Scholar] [CrossRef]
- Ishak, N.A.I.M.; Kamarudin, S.K.; Timmiati, S.N.; Karim, N.A.; Basri, S. Biogenic Platinum from Agricultural Wastes Extract for Improved Methanol Oxidation Reaction in Direct Methanol Fuel Cell. J. Adv. Res. 2021, 28, 63–75. [Google Scholar] [CrossRef]
- Yang, J.; Luo, C.; He, S.; Li, J.; Meng, B.; Zhang, D.; Xue, Z.; Zhou, X.; Lu, X. Synthesis of Three-Dimensional Au-Graphene Quantum Dots@Pt Core–Shell Dendritic Nanoparticles for Enhanced Methanol Electro-Oxidation. Nanotechnology 2019, 30, 495706. [Google Scholar] [CrossRef]
- He, W.; Han, X.; Jia, H.; Cai, J.; Zhou, Y.; Zheng, Z. AuPt Alloy Nanostructures with Tunable Composition and Enzyme-like Activities for Colorimetric Detection of Bisulfide. Sci. Rep. 2017, 7, 40103. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, H.; Wu, P.; Zhang, H.; Zhou, B.; Cai, C. Bimetallic Pt–Au Nanocatalysts Electrochemically Deposited on Graphene and Their Electrocatalytic Characteristics towards Oxygen Reduction and Methanol Oxidation. Phys. Chem. Chem. Phys. 2011, 13, 4083–4094. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Meyer, J.C.; Scardaci, V.; Casiraghi, C.; Lazzeri, M.; Mauri, F.; Piscanec, S.; Jiang, D.; Novoselov, K.S.; Roth, S.; et al. Raman Spectrum of Graphene and Graphene Layers. Phys. Rev. Lett. 2006, 97, 187401. [Google Scholar] [CrossRef] [Green Version]
- Malard, L.M.; Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S. Raman Spectroscopy in Graphene. Phys. Rep. 2009, 473, 51–87. [Google Scholar] [CrossRef]
- Xu, H.; Wu, X.; Li, X.; Luo, C.; Liang, F.; Orignac, E.; Zhang, J.; Chu, J. Properties of Graphene-Metal Contacts Probed by Raman Spectroscopy. Carbon N. Y. 2018, 127, 491–497. [Google Scholar] [CrossRef]
- Wang, W.X.; Liang, S.H.; Yu, T.; Li, D.H.; Li, Y.B.; Han, X.F. The Study of Interaction between Graphene and Metals by Raman Spectroscopy. J. Appl. Phys. 2011, 109, 07C501. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, Y.; Kim, P.; Pinczuk, A. Electric Field Effect Tuning of Electron-Phonon Coupling in Graphene. Phys. Rev. Lett. 2007, 98, 166802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stampfer, C.; Molitor, F.; Graf, D.; Ensslin, K.; Jungen, A.; Hierold, C.; Wirtz, L. Raman Imaging of Doping Domains in Graphene on SiO2. Appl. Phys. Lett. 2007, 91, 241907. [Google Scholar] [CrossRef]
- Das, A.; Pisana, S.; Chakraborty, B.; Piscanec, S.; Saha, S.K.; Waghmare, U.V.; Novoselov, K.S.; Krishnamurthy, H.R.; Geim, A.K.; Ferrari, A.C.; et al. Monitoring Dopants by Raman Scattering in an Electrochemically Top-Gated Graphene Transistor. Nat. Nanotechnol. 2008, 3, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Mukherjee, S.; Gangopadhyay, K.; Gangopadhyay, S. Ultrafine Pt Nanoparticle Induced Doping/Strain of Single Layer Graphene: Experimental Corroboration between Conduction and Raman Characteristics. J. Mater. Sci. Mater. Electron. 2015, 26, 4746–4753. [Google Scholar] [CrossRef]
- Iqbal, M.W.; Iqbal, M.Z.; Khan, M.F.; Jin, X.; Hwang, C.; Eom, J. Modification of the Structural and Electrical Properties of Graphene Layers by Pt Adsorbates. Sci. Technol. Adv. Mater. 2014, 15, 55002. [Google Scholar] [CrossRef]
- Beams, R.; Gustavo Cançado, L.; Novotny, L. Raman Characterization of Defects and Dopants in Graphene. J. Phys. Condens. Matter. 2015, 27, 83002. [Google Scholar] [CrossRef]
- Zhong, X.; Yu, H.; Wang, X.; Liu, L.; Jiang, Y.; Wang, L.; Zhuang, G.; Chu, Y.; Li, X.; Wang, J. Pt@Au Nanorods Uniformly Decorated on Pyridyne Cycloaddition Graphene as a Highly Effective Electrocatalyst for Oxygen Reduction. ACS Appl. Mater. Interfaces 2014, 6, 13448–13454. [Google Scholar] [CrossRef]
- Camci, M.T.; Ulgut, B.; Kocabas, C.; Suzer, S. In-Situ XPS Monitoring and Characterization of Electrochemically Prepared Au Nanoparticles in an Ionic Liquid. ACS Omega 2017, 2, 478–486. [Google Scholar] [CrossRef]
- Sahoo, S.R.; Ke, S.-C. Spin-Orbit Coupling Effects in Au 4f Core-Level Electronic Structures in Supported Low-Dimensional Gold Nanoparticles. Nanomaterials 2021, 11, 554. [Google Scholar] [CrossRef]
- Lu, Q.; Huang, J.; Han, C.; Sun, L.; Yang, X. Facile Synthesis of Composition-Tunable PtRh Nanosponges for Methanol Oxidation Reaction. Electrochim. Acta 2018, 266, 305–311. [Google Scholar] [CrossRef]
- Joshi, V.S.; Poudyal, D.C.; Satpati, A.K.; Patil, K.R.; Haram, S.K. Methanol Oxidation Reaction on Pt Based Electrocatalysts Modified Ultramicroelectrode (UME): Novel Electrochemical Method for Monitoring Rate of CO Adsorption. Electrochim. Acta 2018, 286, 287–295. [Google Scholar] [CrossRef]
- Wang, C.; Yang, F.; Gao, L.; Xu, S.; Fan, L.; Guo, T.; Liu, Y.; Zhou, L.; Zhang, Y. AuPt Nanoparticles Clusters on MWCNTs with Enhanced Electrocatalytic Activity for Methanol Oxidation. Catalysts 2018, 8, 669. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.H.; Krewer, U.; Scott, K. Principles and Materials Aspects of Direct Alkaline Alcohol Fuel Cells. Energies 2010, 3, 1499–1528. [Google Scholar] [CrossRef]
- Roy, S.; Payra, S.; Challagulla, S.; Arora, R.; Roy, S.; Chakraborty, C. Enhanced Photoinduced Electrocatalytic Oxidation of Methanol Using Pt Nanoparticle-Decorated TiO2–Polyaniline Ternary Nanofibers. ACS Omega 2018, 3, 17778–17788. [Google Scholar] [CrossRef] [Green Version]
- Pavlets, A.; Alekseenko, A.; Menshchikov, V.; Belenov, S.; Volochaev, V.; Pankov, I.; Safronenko, O.; Guterman, V. Influence of Electrochemical Pretreatment Conditions of PtCu/C Alloy Electrocatalyst on Its Activity. Nanomaterials 2021, 11, 1499. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Sun, L.; Wang, W.; Chen, Z. Enhanced Electrocatalytic Activity of PtCu Bimetallic Nanoparticles on CeO2/Carbon Nanotubes for Methanol Electro-Oxidation. Int. J. Hydrog. Energy 2020, 45, 8558–8567. [Google Scholar] [CrossRef]
- Lei, W.; Li, M.; He, L.; Meng, X.; Mu, Z.; Yu, Y.; Ross, F.M.; Yang, W. A General Strategy for Bimetallic Pt-Based Nano-Branched Structures as Highly Active and Stable Oxygen Reduction and Methanol Oxidation Bifunctional Catalysts. Nano Res. 2020, 13, 638–645. [Google Scholar] [CrossRef]
- Menshikov, V.S.; Novomlinsky, I.N.; Belenov, S.V.; Alekseenko, A.A.; Safronenko, O.I.; Guterman, V.E. Methanol, Ethanol, and Formic Acid Oxidation on New Platinum-Containing Catalysts. Catalysts 2021, 11, 158. [Google Scholar] [CrossRef]
- Dong, X.; Lu, S.; Xu, W.; Li, S. The Fabrication Composite Material of Bimetallic Micro/Nanostructured Palladium–Platinum Alloy and Graphene on Nickel Foam for the Enhancement of Electrocatalytic Activity. New J. Chem. 2021, 45, 6550–6559. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Q.; Qing, S.; Gao, Z.; Tong, X.; Yang, N. Modulating Electronic Structure of an Au-Nanorod-Core–PdPt-Alloy-Shell Catalyst for Efficient Alcohol Electro-Oxidation. Adv. Energy Mater. 2021, 11, 2100812. [Google Scholar] [CrossRef]
- Ekrami-Kakhki, M.-S.; Naeimi, A.; Donyagard, F. Pt Nanoparticles Supported on a Novel Electrospun Polyvinyl Alcohol-CuOCo3O4/Chitosan Based on Sesbania Sesban Plant as an Electrocatalyst for Direct Methanol Fuel Cells. Int. J. Hydrog. Energy 2019, 44, 1671–1685. [Google Scholar] [CrossRef]
- Dutta, S.; Ray, C.; Sasmal, A.K.; Negishi, Y.; Pal, T. Fabrication of Dog-Bone Shaped Au NRcore–Pt/Pdshell Trimetallic Nanoparticle-Decorated Reduced Graphene Oxide Nanosheets for Excellent Electrocatalysis. J. Mater. Chem. A 2016, 4, 3765–3776. [Google Scholar] [CrossRef]
- Dix, S.T.; Lu, S.; Linic, S. Critical Practices in Rigorously Assessing the Inherent Activity of Nanoparticle Electrocatalysts. ACS Catal. 2020, 10, 10735–10741. [Google Scholar] [CrossRef]
- Zhang, Y.; Chang, G.; Shu, H.; Oyama, M.; Liu, X.; He, Y. Synthesis of Pt–Pd Bimetallic Nanoparticles Anchored on Graphene for Highly Active Methanol Electro-Oxidation. J. Power Sources 2014, 262, 279–285. [Google Scholar] [CrossRef]
- Chang, G.; Cai, Z.; Jia, H.; Zhang, Z.; Liu, X.; Liu, Z.; Zhu, R.; He, Y. High Electrocatalytic Performance of a Graphene-Supported PtAu Nanoalloy for Methanol Oxidation. Int. J. Hydrog. Energy 2018, 43, 12803–12810. [Google Scholar] [CrossRef]
- Xu, L.; Cui, Q.; Zhang, H.; Jiao, A.; Tian, Y.; Li, S.; Li, H.; Chen, M.; Chen, F. Ultra-Clean PtPd Nanoflowers Loaded on GO Supports with Enhanced Low-Temperature Electrocatalytic Activity for Fuel Cells in Harsh Environment. Appl. Surf. Sci. 2020, 511, 145603. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Sun, S.; Wang, L.; Guo, T.; Zhang, D.; Xue, Z.; Zhou, X. PtNi Nanoparticles Supported on Electrochemically Reduced Porous Graphene Oxide for Methanol Oxidation Reaction. Chem. Phys. Lett. 2019, 730, 575–581. [Google Scholar] [CrossRef]
- Yang, X.; Liang, Z.; Chen, S.; Ma, M.; Wang, Q.; Tong, X.; Zhang, Q.; Ye, J.; Gu, L.; Yang, N. A Phosphorus-Doped Ag@Pd Catalyst for Enhanced C-C Bond Cleavage during Ethanol Electrooxidation. Small 2020, 16, 2004727. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | MAu (mg) | Mpt (mg) | MAu/MPt | Average Size of NPs (nm) | VAu (μL) | Vpt (μL) | Au/Pt Atomic Ratio |
|---|---|---|---|---|---|---|---|
| TG/Au60Pt40 | 0.253 | 0.166 | 60/40 | 29.5 | 90 | 60 | 1.53 |
| TG/Au56Pt44 | 0.254 | 0.203 | 56/44 | 38.5 | 90 | 75 | 1.26 |
| TG/Au52Pt48 | 0.258 | 0.241 | 52/48 | 44.5 | 90 | 90 | 1.07 |
| TG/Au42Pt58 | 0.247 | 0.345 | 42/58 | 62.5 | 90 | 120 | 0.72 |
| TG’/Au53Pt47 | 0.478 | 0.412 | 53/47 | 45.8 | 90 | 90 | 1.08 |
| Catalyst | Jpf (mA/cm2) | Eonset (V vs. RHE) | ECSA (m2.g−1) |
|---|---|---|---|
| TG | - | - | - |
| TG/Au0.258 | - | - | - |
| TG/Pt0.241 | 1.28 | 0.74 | 8.5 |
| TG/Au60Pt40 | 3.07 | 0.63 | 18.0 |
| TG/Au56Pt44 | 4.78 | 0.60 | 20.0 |
| TG/Au52Pt48 | 5.87 | 0.59 | 23.7 |
| TG/Au42Pt58 | 7.81 | 0.64 | 25.8 |
| TG’/Au53Pt47 | 12.92 | 0.55 | 40.8 |
| Catalyst | Solutions | Jpf (mA/cm2) | Ref. |
|---|---|---|---|
| TG’/Au53Pt47 | 0.5 M KOH | 12.92 | This work |
| Pt1Pd3NPs/GO | 1 M NaOH | 2.73 | [68] |
| PtAuNA/GO | 1 M NaOH | 7.27 | [69] |
| GO/PtPd | 1 M KOH | 2.59 | [70] |
| Pt52Fe29Co19@GO-7% | 0.5 M H2SO4 | 3.42 | [14] |
| PtNi/GO | 0.5 M H2SO4 | 4.65 | [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, A.T.; Tran, V.V.; Siahaan, A.; Kan, H.-C.; Hsu, Y.-J.; Hsu, C.-C. Free-Standing, Interwoven Tubular Graphene Mesh-Supported Binary AuPt Nanocatalysts: An Innovative and High-Performance Anode Methanol Oxidation Catalyst. Nanomaterials 2022, 12, 1689. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12101689
Nguyen AT, Tran VV, Siahaan A, Kan H-C, Hsu Y-J, Hsu C-C. Free-Standing, Interwoven Tubular Graphene Mesh-Supported Binary AuPt Nanocatalysts: An Innovative and High-Performance Anode Methanol Oxidation Catalyst. Nanomaterials. 2022; 12(10):1689. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12101689
Chicago/Turabian StyleNguyen, An T., Van Viet Tran, Asnidar Siahaan, Hung-Chih Kan, Yung-Jung Hsu, and Chia-Chen Hsu. 2022. "Free-Standing, Interwoven Tubular Graphene Mesh-Supported Binary AuPt Nanocatalysts: An Innovative and High-Performance Anode Methanol Oxidation Catalyst" Nanomaterials 12, no. 10: 1689. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12101689