
Liposomal Form of 2,4-Dinitrophenol Lipophilic Derivatives as a Promising Therapeutic Agent for ATP Synthesis Inhibition

 , , , , , ,
, , , , , ,
Abstract
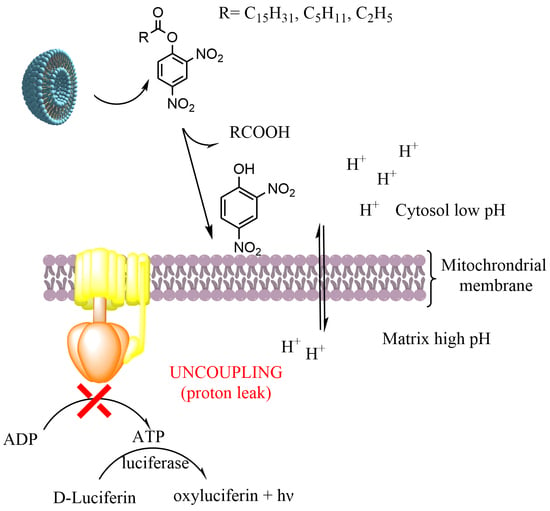
:
1. Introduction
2. Materials and Methods
2.1. Synthesis of 2,4-DNP Carboxylic Acid Esters
2.1.1. Synthesis of 2,4-DNP Palmitic Acid Ester (2,4-DNP-C16) Using the Activated Ester Method
2.1.2. Synthesis of 2,4-DNP-C16 Ester Using Palmitic Acid Chloride
2.1.3. Synthesis of 2,4-Dinitrophenol Caproic Acid Ester (2,4-DNP-C6)
2.1.4. Synthesis of 2,4-Dinitrophenol Propionic Acid Ester (2,4-DNP-C3)
2.2. Gas Chromatography–Mass Spectrometry (GC–MS)
2.3. Octanol–Water Partition Coefficients Measurements (LogP)
2.4. 2,4-DNP Concentration Measurement
2.5. Liposomal Formulation
2.6. Stability Studies of Liposome
2.7. In Vitro Test
2.7.1. Cells
2.7.2. Cytotoxicity Test
2.7.3. Mitopotential Detection
2.7.4. Luciferin–Luciferase Coupled Assay
2.7.5. Measurement of Total ATP
2.8. Statistics
3. Results
3.1. Synthesis of 2,4-DNP Esters
3.2. In Vitro Tests of 2,4-DNP Esters
3.3. Liposomal Formulations of 2,4-DNP Derivatives
3.4. In Vitro Test of Liposomal Formulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, K.C.; Kim, S.H. Interrelationship between fatty liver and insulin resistance in the development of type 2 diabetes. J. Clin. Endocrinol. Metab. 2011, 96, 1093–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Di, F.; Wang, Q.; Shao, J.; Gao, L.; Wang, L.; Li, Q.; Li, N. Non-alcoholic fatty liver disease is a risk factor for the development of diabeticnephropathy in patients with type 2 diabetes mellitus. PLoS ONE 2015, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Li, W.D.; Fu, K.F.; Li, G.M.; Lian, Y.S.; Ren, A.M.; Chen, Y.J.; Xia, J.R. Comparison of effects of obesity and non-alcoholic fatty liver disease on incidence of type 2 diabetes mellitus. World J. Gastroenterol. 2015, 21, 9607–9613. [Google Scholar] [CrossRef] [PubMed]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef]
- Cainelli, F. Liver diseases in developing countries. World J. Hepatol. 2012, 4, 66–67. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Younossi, Y.; Golabi, P.; Mishra, A.; Rafiq, N.; Henry, L. Epidemiology of chronic liver diseases in the USA in the past three decades. Gut 2020, 69, 564–568. [Google Scholar] [CrossRef]
- Smeuninx, B.; Boslem, E.; Febbraio, M.A. Current and Future Treatments in the Fight against Non-Alcoholic Fatty Liver Disease. Cancers 2020, 12, 1714. [Google Scholar] [CrossRef]
- De Zeng, M.; Fan, J.G.; Lu, L.G.; Li, Y.M.; Chen, C.W.; Wang, B.Y.; Mao, Y.M.; Chinese National Consensus Workshop on Nonalcoholic Fatty Liver Disease. Guidelines for the diagnosis and treatment of nonalcoholic fatty liver diseases. J. Dig. Dis. 2008, 9, 108–112. [Google Scholar] [CrossRef]
- Oh, H.; Jun, D.W.; Saeed, W.K.; Nguyen, M.H. Non-alcoholic fatty liver diseases: Update on the challenge of diagnosis and treatment. Clin. Mol. Hepatol. 2016, 22, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Shulman, G.I. Therapeutic potential of mitochondrial uncouplers for the treatment of metabolic associated fatty liver disease and NASH. Mol. Metab. 2021, 46, 101178. [Google Scholar] [CrossRef] [PubMed]
- Goldgof, M.; Xiao, C.; Chanturiya, T.; Jou, W.; Gavrilova, O.; Reitman, M.L. The chemical uncoupler 2,4-dinitrophenol (DNP) protects against diet-induced obesity and improves energy homeostasis in mice at thermoneutrality. J. Biol. Chem. 2014, 289, 19341–19350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lardy, H.A.; Wellman, H. The catalytic effect of 2,4-dinitrophenol on adenosinetriphosphate hydrolysis by cell particles and soluble enzymes. J. Biol. Chem. 1953, 201, 357–370. [Google Scholar] [CrossRef]
- Hua, X.; Du, G.L.; Han, J.; Xu, Y. Bioprocess Intensification for Whole-Cell Catalysis of Catabolized Chemicals with 2,4-Dinitrophenol Uncoupling. ACS Sustain. Chem. Eng. 2020, 8, 15782–15790. [Google Scholar] [CrossRef]
- Childress, E.S.; Alexopoulos, S.J.; Hoehn, K.L.; Santos, W.L. Small Molecule Mitochondrial Uncouplers and Their Therapeutic Potential. J. Med. Chem. 2018, 61, 4641–4655. [Google Scholar] [CrossRef]
- Sousa, D.; Carmo, H.; Roque Bravo, R.; Carvalho, F.; Bastos, M.D.L.; Guedes de Pinho, P.; Dias da Silva, D. Diet aid or aid to die: An update on 2,4-dinitrophenol (2,4-DNP) use as a weight-loss product. Arch. Toxicol. 2020, 94, 1071–1083. [Google Scholar] [CrossRef]
- Perry, R.J.; Kim, T.; Zhang, X.M.; Lee, H.Y.; Pesta, D.; Popov, V.B.; Zhang, D.; Rahimi, Y.; Jurczak, M.J.; Cline, G.W. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 2013, 18, 740–748. [Google Scholar] [CrossRef] [Green Version]
- Perry, R.J.; Zhang, D.; Zhang, X.-M.; Boyer, J.L.; Shulman, G.I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 2015, 347, 1253–1256. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Song, X.; Fu, Y.; Gong, T.; Zhang, Q. Sustained-release mitochondrial protonophore reverses nonalcoholic fatty liver disease in rats. Int. J. Pharm. 2017, 530, 230–238. [Google Scholar] [CrossRef]
- Kim, D.H.; Jahn, A.; Cho, S.J.; Kim, J.S.; Ki, M.H.; Kim, D.D. Lyotropic liquid crystal systems in drug delivery: A review. J. Pharm. Investig. 2015, 45, 1–11. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New Developments in Liposomal Drug Delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Lou, J.; Best, M.D. Strategies for altering lipid self-assembly to trigger liposome cargo release. Chem. Phys. Lipids 2020, 232, 104966. [Google Scholar] [CrossRef]
- Bibi, S.; Lattmann, E.; Mohammed, A.R.; Perrie, Y. Trigger release liposome systems: Local and remote controlled delivery? J. Microencapsul. 2012, 29, 262–276. [Google Scholar] [CrossRef]
- Cipolla, D.; Wu, H.; Gonda, I.; Eastman, S.; Redelmeier, T.; Chan, H.K. Modifying the release properties of liposomes toward personalized medicine. J. Pharm. Sci. 2014, 103, 1851–1862. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Goel, S.; Liu, H.J.; Carter, K.A.; Jiang, D.; Geng, J.; Kutyreff, C.J.; Engle, J.W.; Huang, W.C.; Shao, S. Intrabilayer 64Cu Labeling of Photoactivatable, Doxorubicin-Loaded Stealth Liposomes. ACS Nano 2017, 11, 12482–12491. [Google Scholar] [CrossRef]
- Samuelsson, E.; Shen, H.; Blanco, E.; Ferrari, M.; Wolfram, J. Contribution of Kupffer cells to liposome accumulation in the liver. Colloids Surf. B Biointerfaces 2017, 158, 356–362. [Google Scholar] [CrossRef]
- Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.C. Immunological and toxicological considerations for the design of liposomes. Nanomaterials 2020, 10, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulati, M.; Grover, M.; Singh, S.; Singh, M. Lipophilic drug derivatives in liposomes. Int. J. Pharm. 1998, 165, 129–168. [Google Scholar] [CrossRef]
- Liederer, B.M.; Borchardt, R.T. Enzymes involved in the bioconversion of ester-based prodrugs. J. Pharm. Sci. 2006, 95, 1177–1195. [Google Scholar] [CrossRef] [PubMed]
- Rogerson, M.L.; Robinson, B.H.; Bucak, S.; Walde, P. Kinetic studies of the interaction of fatty acids with phosphatidylcholine vesicles (liposomes). Colloids Surf. B Biointerfaces 2006, 48, 24–34. [Google Scholar] [CrossRef]
- Amézqueta, S.; Subirats, X.; Fuguet, E.; Rosés, M.; Ràfols, C. Chapter 6—Octanol-Water Partition Constant. In Handbooks in Separation Science, Liquid-Phase Extraction; Poole, C.F., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 183–208. [Google Scholar] [CrossRef]
- Stewart, J.C.M. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 1980, 104, 10–14. [Google Scholar] [CrossRef]
- Matta, H.; Gopalakrishnan, R.; Choi, S.; Prakash, R.; Natarajan, V.; Prins, R.; Gong, S.; Chitnis, S.D.; Kahn, M.; Han, X.; et al. Development and characterization of a novel luciferase based cytotoxicity assay. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Baklaushev, V.P.; Kilpeläinen, A.; Petkov, S.; Abakumov, M.A.; Grinenko, N.F.; Yusubalieva, G.M.; Latanova, A.A.; Gubskiy, I.L.; Zabozlaev, F.G.; Starodubova, E.S. Luciferase Expression Allows Bioluminescence Imaging but Imposes Limitations on the Orthotopic Mouse (4T1) Model of Breast Cancer. Sci. Rep. 2017, 7, 7715. [Google Scholar] [CrossRef]
- Torres de Pinedo, A.; Peñalver, P.; Pérez-Victoria, I.; Rondón, D.; Morales, J.C. Synthesis of new phenolic fatty acid esters and their evaluation as lipophilic antioxidants in an oil matrix. Food Chem. 2007, 105, 657–665. [Google Scholar] [CrossRef]
- Appendino, G.; Minassi, A.; Daddario, N.; Bianchi, F.; Tron, G.C. Chemoselective Esterification of Phenolic Acids and Alcohols. Org. Lett. 2002, 4, 3839–3841. [Google Scholar] [CrossRef]
- Shelkov, R.; Nahmany, M.; Melman, A. Selective esterifications of alcohols and phenols through carbodiimide couplings. Org. Biomol. Chem. 2004, 2, 397–401. [Google Scholar] [CrossRef]
- Hosseini Sarvari, M.; Sharghi, H. Zinc oxide (ZnO) as a new, highly efficient, and reusable catalyst for acylation of alcohols, phenols and amines under solvent free conditions. Tetrahedron 2005, 61, 10903–10907. [Google Scholar] [CrossRef]
- Ralston, A.W.; Ingle, A.; McCorkle, M.R. Observations on the effect of some solvents upon the acylation of phenol with high molecular weight acid chlorides. J. Org. Chem. 1942, 07, 457–461. [Google Scholar] [CrossRef]
- Fattahi, N.; Ayubi, M.; Ramazani, A. Amidation and esterification of carboxylic acids with amines and phenols by N,N′-diisopropylcarbodiimide: A new approach for amide and ester bond formation in water. Tetrahedron 2018, 74, 4351–4356. [Google Scholar] [CrossRef]
- Fang, M.; Lei, F.; Zhou, J.; Wu, Y.N.; Gong, Z.Y. Rapid, simple and selective determination of 2,4-dinitrophenol by molecularly imprinted spin column extraction coupled with fluorescence detection. Chin. Chem. Lett. 2014, 25, 1492–1494. [Google Scholar] [CrossRef]
- Misra, A.D.; Agarwal, S.K.; Kumar, S.; Rajput, R.P.S. Thin layer chromatographic separation and identification of some phenols on calcium sulphate: Determination of phloroglucinol and resorcinol. Indian J. Chem. Technol. 1998, 5, 383–386. [Google Scholar] [CrossRef]
- Pillai, I.M.S.; Gupta, A.K. Batch and continuous flow anodic oxidation of 2,4-dinitrophenol: Modeling, degradation pathway and toxicity. J. Electroanal. Chem. 2015, 756, 108–117. [Google Scholar] [CrossRef]
- Shrestha, R.; Johnson, E.; Byrne, F.L. Exploring the therapeutic potential of mitochondrial uncouplers in cancer. Mol. Metab. 2021, 51, 101222. [Google Scholar] [CrossRef]
- Craig, F.F.; Simmonds, A.C.; Watmore, D.; McCapra, F.; White, M.R. Membrane-permeable luciferin esters for assay of firefly luciferase in live intact cell. Biochem. J. 1991, 276, 637–641. [Google Scholar] [CrossRef] [Green Version]
- Stuart McLaughlin. The Mechanism of Action of DNP on Phospholipid Bilayer Membranes. J. Membr. Biol. 1972, 9, 361–372. [Google Scholar] [CrossRef]
- Yang, J.J.; Kim, K.-J.; Lee, V.H.L. Role of P-Glycoprotein in Restricting Propranolol Transport in Cultured Rabbit Conjunctival Epithelial Cell Layers. Pharm. Res. 2000, 17, 533–538. [Google Scholar] [CrossRef]
- Behnel, H.J.; Seydewitz, H.H. Changes of the membrane potential during formation of heat shock puffs induced by ion carriers in Drosophila salivary glands. Exp. Cell Res. 1980, 127, 133–141. [Google Scholar] [CrossRef]
- Kaila, K.; Saarikoski, J. Membrane-potential changes caused by 2,4-DNP and related phenols in resting crayfish axons are not due to uncoupling of mitochondria. Comp. Biochem. Physiol. Part C Comp. Pharmacol. 1981, 69, 235–242. [Google Scholar] [CrossRef]
- Vlasova, K.Y.; Piroyan, A.; Le-Deygen, I.M.; Vishwasrao, H.M.; Ramsey, J.D.; Klyachko, N.L.; Golovin, Y.I.; Rudakovskaya, P.G.; Kireev, I.I.; Kabanov, A.V.; et al. Magnetic liposome design for drug release systems responsive to super-low frequency alternating current magnetic field (AC MF). J. Colloid Interface Sci. 2019, 552, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Lindblom, G.; Orädd, G. Lipid lateral diffusion and membrane heterogeneity. Biochim. Biophys. Acta Biomembr. 2009, 1788, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; McEwen, M.L.; Nottingham, S.A.; Maragos, W.F.; Dragicevic, N.B.; Sullivan, P.G.; Springer, J.E. The Mitochondrial Uncoupling Agent 2,4-Dinitrophenol Improves Mitochondrial Function, Attenuates Oxidative Damage, and Increases White Matter Sparing in the Contused Spinal Cord. J. Neurotrauma 2004, 21, 1396–1404. [Google Scholar] [CrossRef]
- Skulachev, V.P. Uncoupling: New approaches to an old problem of bioenergetics. Biochim. Biophys. Acta 1998, 1363, 100–124. [Google Scholar] [CrossRef] [Green Version]
- Demel, R.A.; Kinsky, S.C.; Kinsky, C.B.; Van Deenen, L.L. Effects of temperature and cholesterol on the glucose permeability of liposomes prepared with natural and synthetic lecithins. Biochim. Biophys. Acta (BBA) Biomembr. 1968, 150, 655–665. [Google Scholar] [CrossRef]
- Hays, L.M.; Crowe, J.H.; Wolkers, W.; Rudenko, S. Factors affecting leakage of trapped solutes from phospholipid vesicles during thermotropic phase transitions. Cryobiology 2001, 42, 88–102. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent Name | Added Amount |
|---|---|
| Caproic acid | 686 μL, 5.4 mmol |
| Oxalyl chloride | 703 μL, 8.2 mmol |
| DMF | 12.5 μL, 0.16 mmol |
| Chloroform (caproic acid chloride synthesis) | 10 mL |
| 2,4-DNP | 1 g, 5.4 mmol |
| Triethylamine | 730 μL, 5.4 mmol |
| Chloroform (ester synthesis) | 25 mL |
| Reagent Name | Added Amount |
|---|---|
| Propionic acid | 150 μL, 2 mmol |
| Oxalyl chloride | 257 μL, 3 mmol |
| DMF | 4.6 μL, 0.06 mmol |
| Chloroform (propionic acid chloride synthesis) | 10 mL |
| 2,4-DNP | 368 mg, 2 mmol |
| Triethylamine | 257 μL, 2 mmol |
| Chloroform (ester synthesis) | 25 mL |
| Molecule | Yield, % | LogPoct |
|---|---|---|
| 2,4-DNP (compound 1) | - | 1.61 ± 0.30 |
| 2,4-DNP-C16 (compound 2) | 73 | 5.18 ± 0.79 |
| 2,4-DNP-C6 (compound 3) | 70.5 | 3.07 ± 0.31 |
| 2,4-DNP-C3 (compound 4) | 78 | 1.91 ± 0.24 |
| Loaded Molecule | Lipid Composition (mol/mol %) | Hydrodynamic Diameter, nm *, ** | PDI *, ** | Loading Capacity (mol%) ** |
|---|---|---|---|---|
| 2,4-DNP (compound 1) | DSPC/DSPE-PEG (95/5) | 158 ± 6 | 0.126 ± 0.015 | 1.2 ± 0.3 |
| 2,4-DNP-C3 (compound 4) | DSPC/DSPE-PEG/2,4-DNP-C3 (82/4/14) | 148 ± 1 | 0.144 ± 0.099 | 0.6 ± 0.4 |
| 2,4-DNP-C6 (compound 3) | DSPC/DSPE-PEG/2,4-DNP-C6 (82/4/14) | 160 ± 6 | 0.234 ± 0.010 | 4.0 ± 0.6 |
| 2,4-DNP-C16 (compound 2) | DSPC/DSPE-PEG/2,4-DNP-C16 (82/4/14) | 151 ± 3 | 0.098 ± 0.007 | 3.1 ± 1.2 |
| 2,4-DNP (compound 1) | eggPC/DSPE-PEG (95/5) | 162 ± 5 | 0.117 ± 0.015 | 1.1 ± 0.4 |
| 2,4-DNP-C3 (compound 4) | eggPC/DSPE-PEG/2,4-DNP-C3 (82/4/14) | 182 ± 15 | 0.201 ± 0.123 | 1.4 ± 0.3 |
| 2,4-DNP-C6 (compound 3) | eggPC/DSPE-PEG/2,4-DNP-C6 (82/4/14) | 137 ± 5 | 0.168 ± 0.016 | 4.7 ± 1.1 |
| 2,4-DNP-C16 (compound 2) | eggPC/DSPE-PEG/2,4-DNP-C16 (82/4/14) | 173 ± 9 | 0.251 ± 0.023 | 4.2 ± 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlasova, K.Y.; Ostroverkhov, P.; Vedenyapina, D.; Yakimova, T.; Trusova, A.; Lomakina, G.Y.; Vodopyanov, S.S.; Grin, M.; Klyachko, N.; Chekhonin, V.; et al. Liposomal Form of 2,4-Dinitrophenol Lipophilic Derivatives as a Promising Therapeutic Agent for ATP Synthesis Inhibition. Nanomaterials 2022, 12, 2162. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12132162
Vlasova KY, Ostroverkhov P, Vedenyapina D, Yakimova T, Trusova A, Lomakina GY, Vodopyanov SS, Grin M, Klyachko N, Chekhonin V, et al. Liposomal Form of 2,4-Dinitrophenol Lipophilic Derivatives as a Promising Therapeutic Agent for ATP Synthesis Inhibition. Nanomaterials. 2022; 12(13):2162. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12132162
Chicago/Turabian StyleVlasova, Kseniya Yu., Petr Ostroverkhov, Daria Vedenyapina, Tamara Yakimova, Alla Trusova, Galina Yurievna Lomakina, Stepan Sergeevich Vodopyanov, Mikhail Grin, Natalia Klyachko, Vladimir Chekhonin, and et al. 2022. "Liposomal Form of 2,4-Dinitrophenol Lipophilic Derivatives as a Promising Therapeutic Agent for ATP Synthesis Inhibition" Nanomaterials 12, no. 13: 2162. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12132162