
Comparison of Plasma Deposition of Carbon Nanomaterials Using Various Polymer Materials as a Carbon Atom Source



Abstract
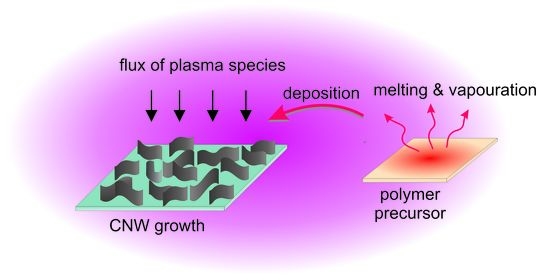
:
1. Introduction
2. Materials and Methods
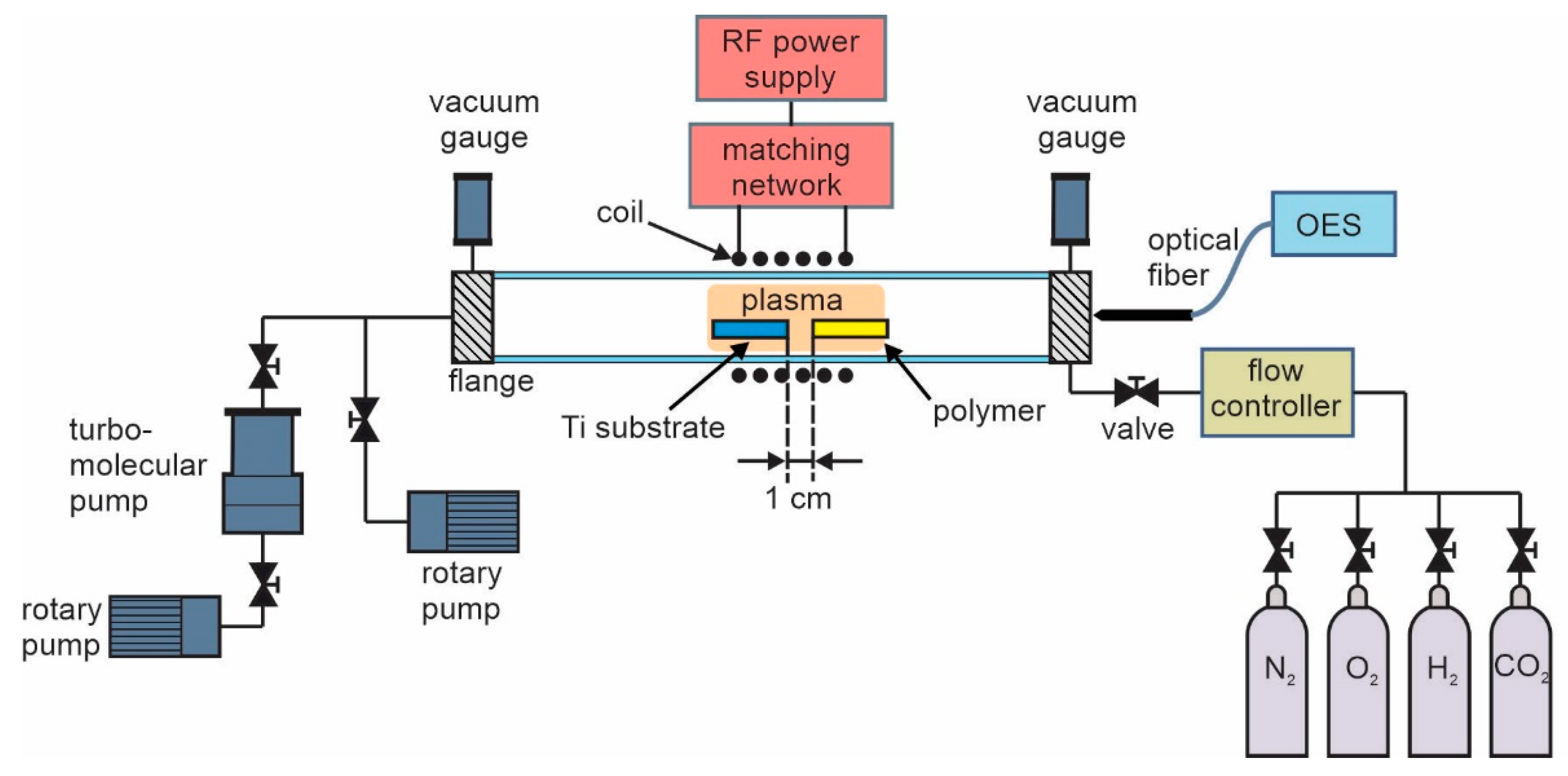
2.1. Plasma Synthesis of Carbon Nanomaterials
2.2. Characterization of the Samples
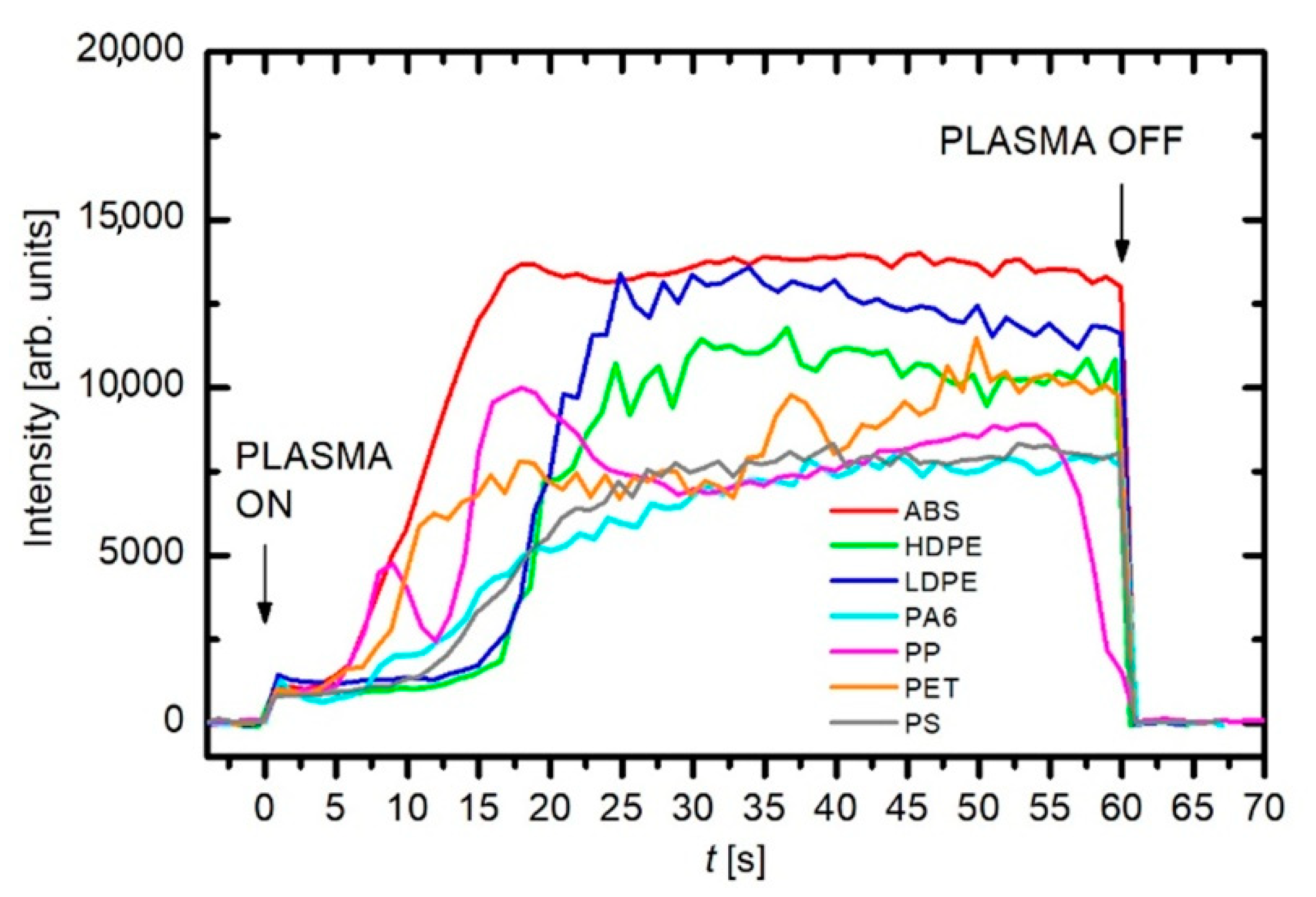
3. Results and Discussion
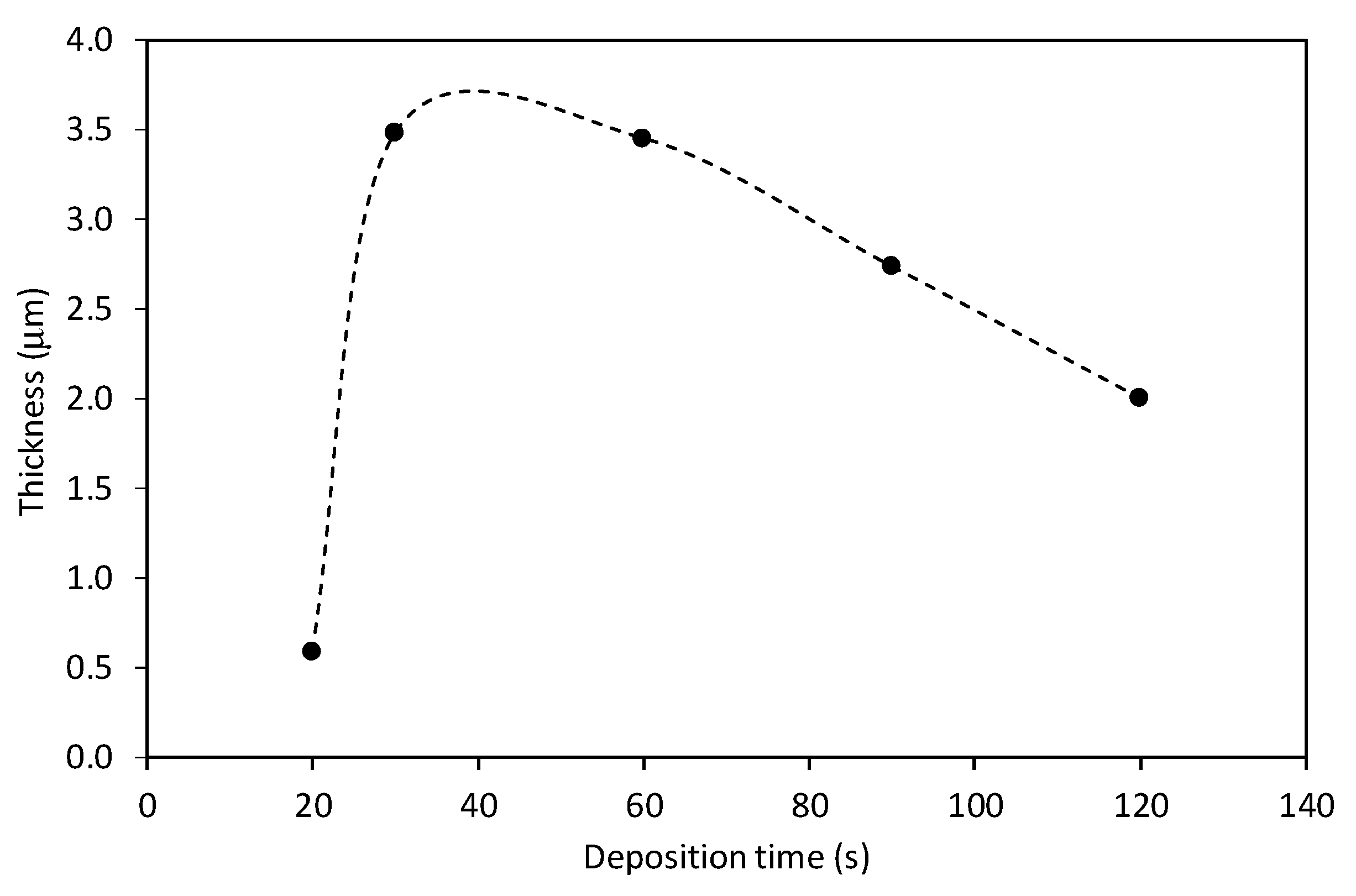
3.1. Influence of the Deposition Time
3.2. Influence of the Polymer Material as a Carbon Atom Source

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Melting T | Decomposition T | Thermal Degradation Products | Refs. |
|---|---|---|---|---|
| PS | 503 K | 549 K | monomer styrene (40%), dimer, trimer, tetramer, pentamer, benzene, ethylbenzene, α-methylstyrene; in the presence of oxygen: phenol, ketones, benzoic acid, benzyl alcohol, benzaldehyde | [14,15,16] |
| PE | 378–408 K | 650 K | propene (up to 25%), propane, ethene, ethane, butene, hexene (formation of the transition state six-membered ring) | [14,15,16] |
| PP | 443 K | 624 K | pentane (24%), 2-methyl-1-pentene, 2,4-dimethyl-1-heptene, propane | [14,15,16] |
| PET | 523–533 K | 698 K | cleavage of ester groups and formation of carboxylic acids and vinyl esters (benzoic acid (43%), acetaldehyde (16%), CO2, vinyl esters of benzoic acid), anhydride containing oligomers, cyclic oligomers; also scission through a six-membered ring transition state; formation of non-volative residues of interconnected aromatic rings was also reported. | [15,16,17,18] |
| PA6 | 498–508 K | 708 K | cyclic oligomers, caprolactam (73%), CO2 | [19] |
| ABS | 383–398 K | 693 K | degradation to its constituents, depending on temperature: butadiene, sytrene, ethylbenzene, N-containing products | [27,28] |
3.3. Influence of the Processing Gas
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, J.H.; Zhu, M.J.; An, Z.L.; Wang, Z.Q.; Toda, M.; Ono, T. Constructing in-chip micro-supercapacitors of 3d graphene nanowall/ruthenium oxides electrode through silicon-based microfabrication technique. J. Power Sources 2018, 401, 204–212. [Google Scholar] [CrossRef]
- Liu, L.L.; Guan, T.; Fang, L.; Wu, F.; Lu, Y.; Luo, H.J.; Song, X.F.; Zhou, M.; Hu, B.S.; Wei, D.P.; et al. Self-supported 3d nico-ldh/gr composite nanosheets array electrode for high-performance supercapacitor. J. Alloys Compd. 2018, 763, 926–934. [Google Scholar] [CrossRef]
- Shin, S.C.; Yoshimura, A.; Matsuo, T.; Mori, M.; Tanimura, M.; Ishihara, A.; Ota, K.; Tachibana, M. Carbon nanowalls as platinum support for fuel cells. J. Appl. Phys. 2011, 110, 104308. [Google Scholar] [CrossRef]
- Krivchenko, V.A.; Itkis, D.M.; Evlashin, S.A.; Semenenko, D.A.; Goodilin, E.A.; Rakhimov, A.T.; Stepanov, A.S.; Suetin, N.V.; Pilevsky, A.A.; Voronin, P.V. Carbon nanowalls decorated with silicon for lithium-ion batteries. Carbon 2012, 50, 1438–1442. [Google Scholar] [CrossRef]
- Takeuchi, W.; Kondo, H.; Obayashi, T.; Hiramatsu, M.; Hori, M. Electron field emission enhancement of carbon nanowalls by plasma surface nitridation. Appl. Phys. Lett. 2011, 98, 123107. [Google Scholar] [CrossRef]
- Wei, W.; Hu, Y.H. Highly conductive na-embedded carbon nanowalls for hole-transport-material-free perovskite solar cells without metal electrodes. J. Mater. Chem. A 2017, 5, 24126–24130. [Google Scholar] [CrossRef]
- Vesel, A.; Zaplotnik, R.; Primc, G.; Mozetič, M. Synthesis of vertically oriented graphene sheets or carbon nanowalls—Review and challenges. Materials 2019, 12, 2968. [Google Scholar] [CrossRef] [Green Version]
- Vesel, A.; Zaplotnik, R.; Primc, G.; Mozetič, M. A review of strategies for the synthesis of n-doped graphene-like materials. Nanomaterials 2020, 10, 2286. [Google Scholar] [CrossRef]
- Vesel, A.; Zaplotnik, R.; Primc, G.; Pirker, L.; Mozetič, M. One-step plasma synthesis of nitrogen-doped carbon nanomesh. Nanomaterials 2021, 11, 837. [Google Scholar] [CrossRef]
- Arnold, C., Jr. Stability of high-temperature polymers. J. Polym. Sci. Macromol. Rev. 1979, 14, 265–378. [Google Scholar] [CrossRef]
- Vohlídal, J. Polymer degradation: A short review. Chem. Teach. Int. 2021, 3, 213–220. [Google Scholar] [CrossRef]
- Dorai, R.; Kushner, M.J. A model for plasma modification of polypropylene using atmospheric pressure discharges. J. Phys. D Appl. Phys. 2003, 36, 666–685. [Google Scholar] [CrossRef]
- Zhang, M.; Buekens, A.; Jiang, X.; Li, X. Dioxins and polyvinylchloride in combustion and fires. Waste Manag. Res. 2015, 33, 630–643. [Google Scholar] [CrossRef]
- Peterson, J.D.; Vyazovkin, S.; Wight, C.A. Kinetics of the thermal and thermo-oxidative degradation of polystyrene, polyethylene and poly(propylene). Macromol. Chem. Phys. 2001, 202, 775–784. [Google Scholar] [CrossRef]
- Gewert, B.; Plassmann, M.M.; MacLeod, M. Pathways for degradation of plastic polymers floating in the marine environment. Environ. Sci. Process. Impacts. 2015, 17, 1513–1521. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, A.; Stec, A.A.; Hull, T.R. Thermal decomposition of polymeric materials. In Sfpe Handbook of Fire Protection Engineering; Hurley, M.J., Gottuk, D., Hall, J.R., Harada, K., Kuligowski, E., Puchovsky, M., Torero, J., Watts, J.M., Wieczorek, C., Eds.; Springer: New York, NY, USA, 2016; pp. 167–254. [Google Scholar]
- Samperi, F.; Puglisi, C.; Alicata, R.; Montaudo, G. Thermal degradation of poly(ethylene terephthalate) at the processing temperature. Polym. Degrad. Stab. 2004, 83, 3–10. [Google Scholar] [CrossRef]
- Holland, B.J.; Hay, J.N. The thermal degradation of pet and analogous polyesters measured by thermal analysis–fourier transform infrared spectroscopy. Polymer 2002, 43, 1835–1847. [Google Scholar] [CrossRef]
- Davis, R.D.; Gilman, J.W.; VanderHart, D.L. Processing degradation of polyamide 6/montmorillonite clay nanocomposites and clay organic modifier. Polym. Degrad. Stab. 2003, 79, 111–121. [Google Scholar] [CrossRef]
- Teii, K.; Shimada, S.; Nakashima, M.; Chuang, A.T.H. Synthesis and electrical characterization of n-type carbon nanowalls. J. Appl. Phys. 2009, 106, 084303. [Google Scholar] [CrossRef]
- Yu, K.H.; Wang, P.X.; Lu, G.H.; Chen, K.H.; Bo, Z.; Chen, J.H. Patterning vertically oriented graphene sheets for nanodevice applications. J. Phys. Chem. Lett. 2011, 2, 537–542. [Google Scholar] [CrossRef]
- Meško, M.; Vretenár, V.; Kotrusz, P.; Hulman, M.; Šoltýs, J.; Skákalová, V. Carbon nanowalls synthesis by means of atmospheric dcpecvd method. Phys. Status Solidi B 2012, 249, 2625–2628. [Google Scholar] [CrossRef]
- Teii, K.; Hori, M.; Goto, T. Negative bias dependence of sulfur and fluorine incorporation in diamond films etched by an sf6 plasma. J. Electrochem. Soc. 2001, 148, G55. [Google Scholar] [CrossRef]
- Lehmann, K.; Yurchenko, O.; Urban, G. Effect of the aromatic precursor flow rate on the morphology and properties of carbon nanostructures in plasma enhanced chemical vapor deposition. RSC Adv. 2016, 6, 32779–32788. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-C.; Bagley, J.D.; Teague, M.L.; Tseng, W.-S.; Yang, K.L.; Zhang, Y.; Li, Y.; Li, Y.; Tour, J.M.; Yeh, N.C. High-yield single-step catalytic growth of graphene nanostripes by plasma enhanced chemical vapor deposition. Carbon 2018, 129, 527–536. [Google Scholar] [CrossRef]
- Ostrikov, K.; Neyts, E.C.; Meyyappan, M. Plasma nanoscience: From nano-solids in plasmas to nano-plasmas in solids. Adv. Phys. 2013, 62, 113–224. [Google Scholar] [CrossRef]
- Suzuki, M.; Wilkie, C.A. The thermal degradation of acrylonitrile-butadiene-styrene terpolymei as studied by tga/ftir. Polym. Degrad. Stab. 1995, 47, 217–221. [Google Scholar] [CrossRef]
- Lee, K.-H.; Shin, D.-H.; Seo, Y.-H. Thermal degradation of nitrogen-containing polymers, acrylonitrile-butadiene-styrene and styrene-acrylonitrile. Korean J. Chem. Eng. 2006, 23, 224–229. [Google Scholar] [CrossRef]
- Hiramatsu, M.; Kondo, H.; Hori, M. Graphene nanowalls. In New Progress on Graphene Research; Gong, J.R., Ed.; IntechOpen: London, UK, 2013. [Google Scholar]
- Cui, L.; Chen, J.; Yang, B.; Sun, D.; Jiao, T. Rf-pecvd synthesis of carbon nanowalls and their field emission properties. Appl. Surf. Sci. 2015, 357, 1–7. [Google Scholar] [CrossRef]
- Jiang, L.; Yang, T.; Liu, F.; Dong, J.; Yao, Z.; Shen, C.; Deng, S.; Xu, N.; Liu, Y.; Gao, H.-J. Controlled synthesis of large-scale, uniform, vertically standing graphene for high-performance field emitters. Adv. Mater. 2013, 25, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, M.; Outlaw, R.A.; Zhao, X.; Manos, D.M.; Holloway, B.C. Synthesis of carbon nanosheets by inductively coupled radio-frequency plasma enhanced chemical vapor deposition. Carbon 2004, 42, 2867–2872. [Google Scholar] [CrossRef]
- Hiramatsu, M.; Hori, M. Carbon Nanowalls: Synthesis and Emerging Applications; Springer: Wien, Austria, 2010. [Google Scholar]
- Gonzalez, E.; Barankin, M.D.; Guschl, P.C.; Hicks, R.F. Ring opening of aromatic polymers by remote atmospheric-pressure plasma. IEEE Trans. Plasma Sci. 2009, 37, 823–831. [Google Scholar] [CrossRef]
- Liang, Y.; Li, J.; Xue, Y.; Tan, T.; Jiang, Z.; He, Y.; Shangguan, W.; Yang, J.; Pan, Y. Benzene decomposition by non-thermal plasma: A detailed mechanism study by synchrotron radiation photoionization mass spectrometry and theoretical calculations. J. Hazard. Mater. 2021, 420, 126584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, S.; Lu, Z.; Chen, X.; Chen, Q.; Gao, P.; Yu, T.; Peng, Z.; Ye, J. Efficient and controllable growth of vertically oriented graphene nanosheets by mesoplasma chemical vapor deposition. Carbon 2019, 147, 341–347. [Google Scholar] [CrossRef]








| Polymer | Structure |
|---|---|
| PS |  |
| LDPE/HDPE | –CH2–CH2– |
| PP |  |
| PET |  |
| PA6 |  |
| ABS |  |
| Polymer | Gas | C (atom. %) | N (atom. %) | O (atom. %) | N/C |
|---|---|---|---|---|---|
| PS | N2 | 93.8 | 3.7 | 2.6 | 0.04 |
| PET | N2 | 94.4 | 2.6 | 3.0 | 0.03 |
| ABS | N2 | 98.0 | 0.7 | 1.3 | 0.03 |
| PA6 | N2 | 94.4 | 2.8 | 2.8 | 0.003 |
| LDPE | N2 | 98.6 | 0.3 | 1.1 | 0.02 |
| HDPE | N2 | 95.6 | 1.7 | 2.8 | 0.01 |
| PP | N2 | 96.7 | 1.0 | 2.3 | 0.01 |
| Polymer | Gas | C (atom. %) | N (atom. %) | O (atom. %) |
|---|---|---|---|---|
| PP | N2 | 96.7 | 1.0 | 2.3 |
| PP | O2 | 97.5 | 2.5 | |
| PP | H2 | 98.3 | 1.7 | |
| PP | CO2 | 98.5 | 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vesel, A.; Zaplotnik, R.; Primc, G.; Paul, D.; Mozetič, M. Comparison of Plasma Deposition of Carbon Nanomaterials Using Various Polymer Materials as a Carbon Atom Source. Nanomaterials 2022, 12, 246. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12020246
Vesel A, Zaplotnik R, Primc G, Paul D, Mozetič M. Comparison of Plasma Deposition of Carbon Nanomaterials Using Various Polymer Materials as a Carbon Atom Source. Nanomaterials. 2022; 12(2):246. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12020246
Chicago/Turabian StyleVesel, Alenka, Rok Zaplotnik, Gregor Primc, Domen Paul, and Miran Mozetič. 2022. "Comparison of Plasma Deposition of Carbon Nanomaterials Using Various Polymer Materials as a Carbon Atom Source" Nanomaterials 12, no. 2: 246. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12020246