
Sunlight-Powered Reverse Water Gas Shift Reaction Catalysed by Plasmonic Au/TiO2 Nanocatalysts: Effects of Au Particle Size on the Activity and Selectivity
 , , , , , , and
, , , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results & Discussion
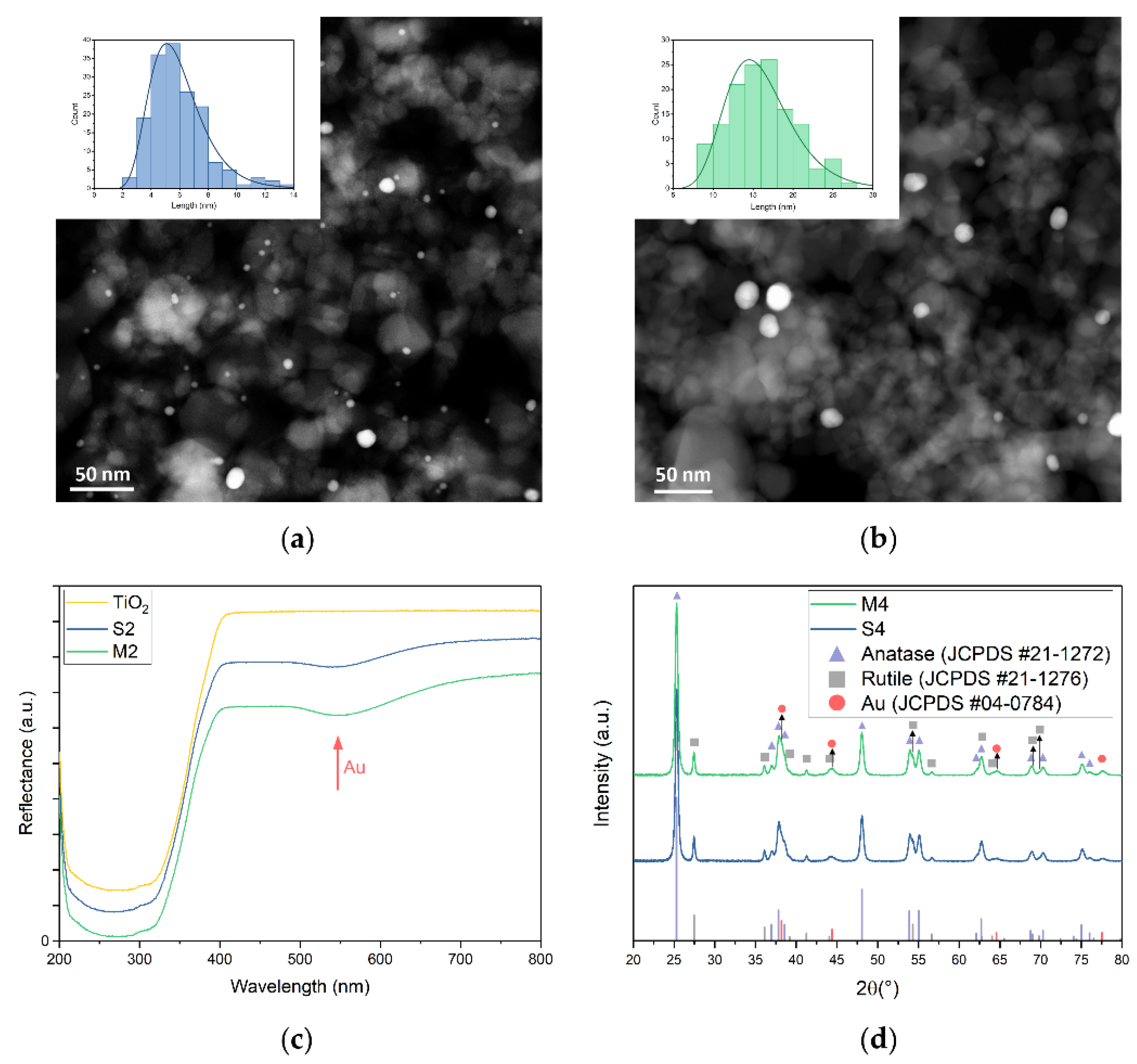
3.1. Catalyst Preparation and Characterisation
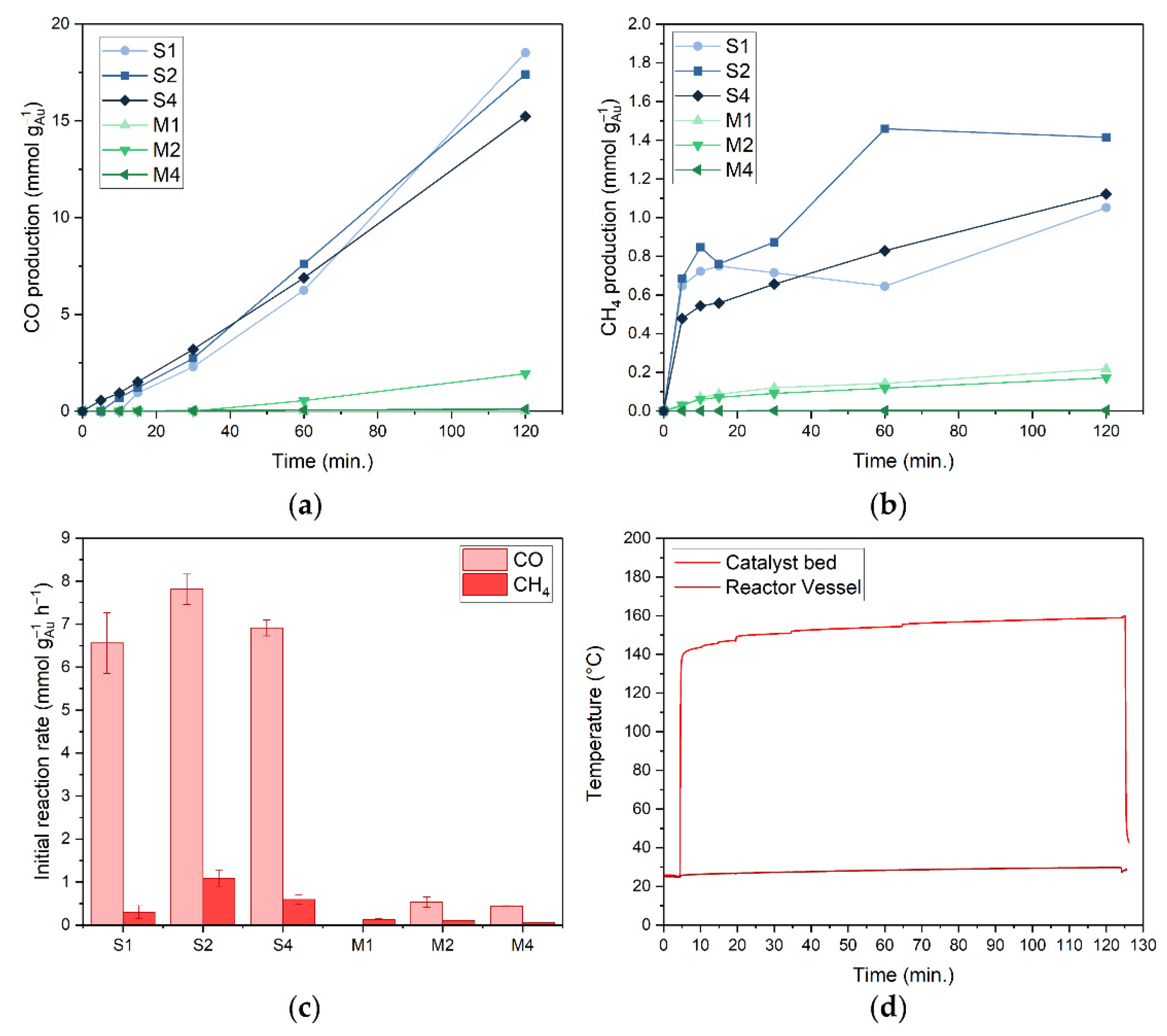
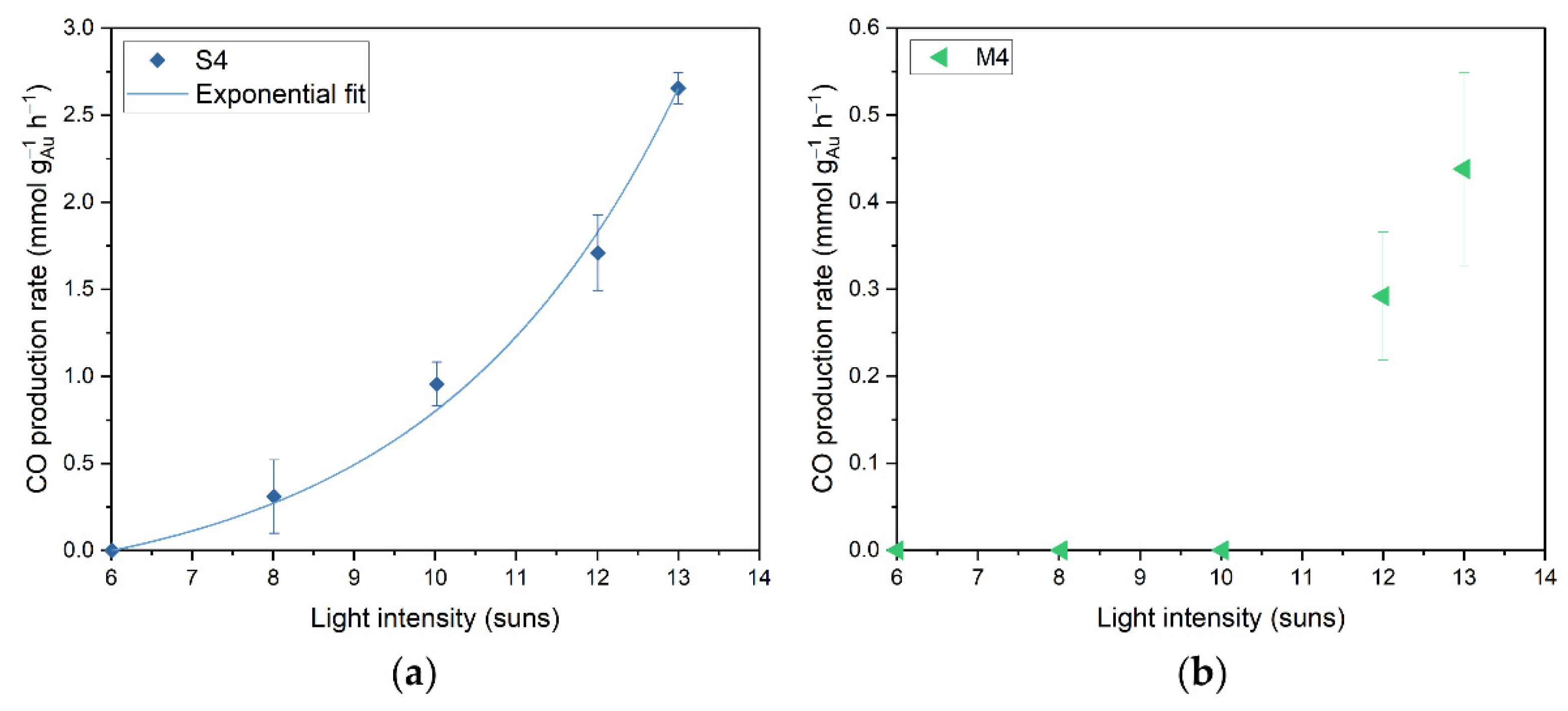
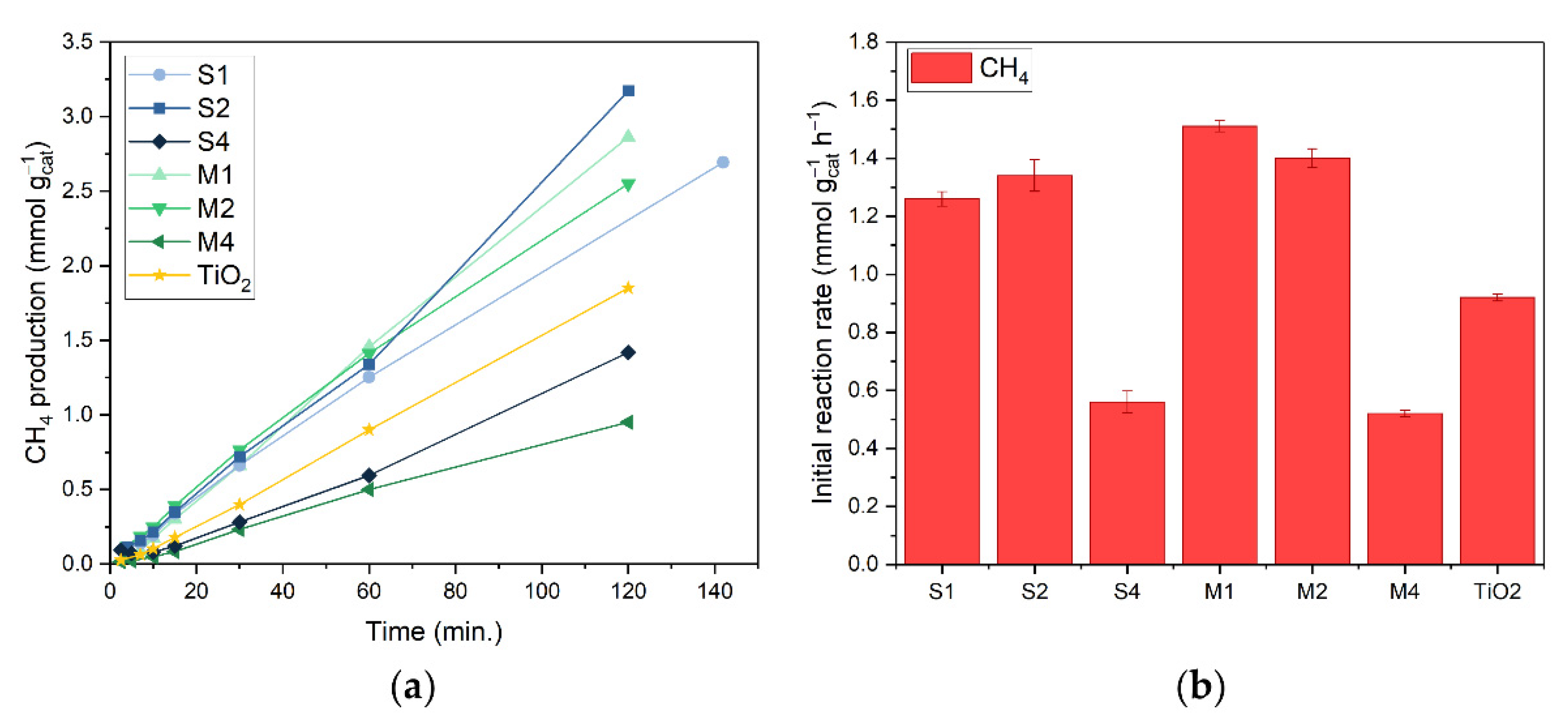
3.2. Catalytic Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IEA. Key World Energy Statistics 2021. Available online: https://www.iea.org/reports/key-world-energy-statistics-2021 (accessed on 1 August 2022).
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported Catalysts for CO2 Methanation: A Review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Gao, W.; Gao, R.; Zhao, Y.; Peng, M.; Song, C.; Li, M.; Li, S.; Liu, J.; Li, W.; Deng, Y.; et al. Photo-Driven Syngas Conversion to Lower Olefins over Oxygen-Decorated Fe5C2 Catalyst. Chem 2018, 4, 2917–2928. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, P.; Unde, R.B.; Kern, C.; Jess, A. Production of Liquid Hydrocarbons with CO2 as Carbon Source based on Reverse Water-Gas Shift and Fischer-Tropsch Synthesis. Chem. Ing. Tech. 2013, 85, 489–499. [Google Scholar] [CrossRef]
- Su, X.; Yang, X.; Zhao, B.; Huang, Y. Designing of highly selective and high-temperature endurable RWGS heterogeneous catalysts:recent advances and the future directions. J. Energy Chem. 2017, 26, 854–867. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Kim, S.S.; Lee, H.H.; Hong, S.C. A study on the effect of support’s reducibility on the reverse water-gas shift reaction over Pt catalysts. Appl. Catal. A Gen. 2012, 423–424, 100–107. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Jaroniec, M.; Chen, X. Cocatalysts for Selective Photoreduction of CO2 into Solar Fuels. Chem. Rev. 2019, 119, 3962–4179. [Google Scholar] [CrossRef]
- Gellé, A.; Moores, A. Plasmonic nanoparticles: Photocatalysts with a bright future. Curr. Opin. Green Sustain. Chem. 2019, 15, 60–66. [Google Scholar] [CrossRef]
- Moores, A.; Goettmann, F. The plasmon band in noble metal nanoparticles: An introduction to theory and applications. New J. Chem. 2006, 30, 1121–1132. [Google Scholar] [CrossRef]
- Nakata, K.; Fujishima, A. TiO2 photocatalysis: Design and applications. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 169–189. [Google Scholar] [CrossRef]
- Verbruggen, S.W. TiO2 photocatalysis for the degradation of pollutants in gas phase: From morphological design to plasmonic enhancement. J. Photochem. Photobiol. C Photochem. Rev. 2015, 24, 64–82. [Google Scholar] [CrossRef]
- Mendonça, C.D.; Khan, S.U.; Rahemi, V.; Verbruggen, S.W.; Machado, S.A.S.; De Wael, K. Surface plasmon resonance-induced visible light photocatalytic TiO2 modified with AuNPs for the quantification of hydroquinone. Electrochim. Acta 2021, 389, 138734. [Google Scholar] [CrossRef]
- Omranian, S.R.; Geluykens, M.; Van Hal, M.; Hasheminejad, N.; Segundo, I.R.; Pipintakos, G.; Denys, S.; Tytgat, T.; Freitas, E.F.; Carneiro, J.; et al. Assessing the potential of application of titanium dioxide for photocatalytic degradation of deposited soot on asphalt pavement surfaces. Constr. Build. Mater. 2022, 350, 128859. [Google Scholar] [CrossRef]
- Van Hal, M.; Verbruggen, S.W.; Yang, X.-Y.; Lenaerts, S.; Tytgat, T. Image analysis and in situ FTIR as complementary detection tools for photocatalytic soot oxidation. Chem. Eng. J. 2019, 367, 269–277. [Google Scholar] [CrossRef]
- Keulemans, M.; Verbruggen, S.W.; Hauchecorne, B.; Martens, J.A.; Lenaerts, S. Activity versus selectivity in photocatalysis: Morphological or electronic properties tipping the scale. J. Catal. 2016, 344, 221–228. [Google Scholar] [CrossRef]
- Meire, M.; Verbruggen, S.W.; Lenaerts, S.; Lommens, P.; Van Der Voort, P.; Van Driessche, I. Microwave-assisted synthesis of mesoporous titania with increased crystallinity, specific surface area, and photocatalytic activity. J. Mater. Sci. 2016, 51, 9822–9829. [Google Scholar] [CrossRef]
- Papavasiliou, A.; Van Everbroeck, T.; Blonda, C.; Oliani, B.; Sakellis, E.; Cool, P.; Canu, P.; Katsaros, F.K. Mesoporous CuO/TiO2 catalysts prepared by the ammonia driven deposition precipitation method for CO preferential oxidation: Effect of metal loading. Fuel 2022, 311, 122491. [Google Scholar] [CrossRef]
- Šuligoj, A.; Arčon, I.; Mazaj, M.; Dražić, G.; Arčon, D.; Cool, P.; Štangar, U.L.; Tušar, N.N. Surface modified titanium dioxide using transition metals: Nickel as a winning transition metal for solar light photocatalysis. J. Mater. Chem. A 2018, 6, 9882–9892. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.-T.; Han, J.-K.; Gain, A.K.; Lee, K.-H.; Saito, F. TEM microstructure characterization of nano TiO2 coated on nano ZrO2 powders and their photocatalytic activity. Mater. Lett. 2006, 60, 2101–2104. [Google Scholar] [CrossRef]
- Wafi, M.A.E.; Ahmed, M.A.; Abdel-Samad, H.S.; Medien, H.A.A. Exceptional removal of methylene blue and p-aminophenol dye over novel TiO2/RGO nanocomposites by tandem adsorption-photocatalytic processes. Mater. Sci. Energy Technol. 2022, 5, 217–231. [Google Scholar] [CrossRef]
- Zeshan, M.; Bhatti, I.A.; Mohsin, M.; Iqbal, M.; Amjed, N.; Nisar, J.; AlMasoud, N.; Alomar, T.S. Remediation of pesticides using TiO2 based photocatalytic strategies: A review. Chemosphere 2022, 300, 134525. [Google Scholar] [CrossRef] [PubMed]
- Burova, D.; Rohlfs, J.; Sastre, F.; Molina, P.M.; Meulendijks, N.; Verheijen, M.A.; Kelchtermans, A.-S.; Elen, K.; Hardy, A.; Van Bael, M.K.; et al. Comparing the Performance of Supported Ru Nanocatalysts Prepared by Chemical Reduction of RuCl3 and Thermal Decomposition of Ru3(CO)12 in the Sunlight-Powered Sabatier Reaction. Catalysts 2022, 12, 284. [Google Scholar] [CrossRef]
- Grote, R.; Habets, R.; Rohlfs, J.; Sastre, F.; Meulendijks, N.; Xu, M.; Verheijen, M.A.; Elen, K.; Hardy, A.; Van Bael, M.K.; et al. Collective photothermal effect of Al2O3-supported spheroidal plasmonic Ru nanoparticle catalysts in the sunlight-powered Sabatier reaction. ChemCatChem 2020, 12, 5618–5622. [Google Scholar] [CrossRef]
- Sastre, F.; Puga, A.V.; Liu, L.; Corma, A.; García, H. Complete Photocatalytic Reduction of CO2 to Methane by H2 under Solar Light Irradiation. J. Am. Chem. Soc. 2014, 136, 6798–6801. [Google Scholar] [CrossRef]
- Sastre, F.; Versluis, C.; Meulendijks, N.; Rodríguez-Fernández, J.; Sweelssen, J.; Elen, K.; Van Bael, M.K.; den Hartog, T.; Verheijen, M.A.; Buskens, P. Sunlight-Fueled, Low-Temperature Ru-Catalyzed Conversion of CO2 and H2 to CH4 with a High Photon-to-Methane Efficiency. ACS Omega 2019, 4, 7369–7377. [Google Scholar] [CrossRef]
- Wang, Z.-j.; Song, H.; Liu, H.; Ye, J. Coupling of Solar Energy and Thermal Energy for Carbon Dioxide Reduction: Status and Prospects. Angew. Chem. Int. Ed. 2020, 59, 8016–8035. [Google Scholar] [CrossRef]
- van der Zwaan, B.; Detz, R.; Meulendijks, N.; Buskens, P. Renewable natural gas as climate-neutral energy carrier? Fuel 2022, 311, 122547. [Google Scholar] [CrossRef]
- Rohlfs, J.; Bossers, K.W.; Meulendijks, N.; Mackenzie, F.V.; Xu, M.; Verheijen, M.A.; Buskens, P.; Sastre, F. Continuous-Flow Sunlight-Powered CO2 Methanation Catalyzed by γ-Al2O3-Supported Plasmonic Ru Nanorods. Catalysts 2022, 12, 126. [Google Scholar] [CrossRef]
- Borah, R.; Verbruggen, S.W. Effect of size distribution, skewness and roughness on the optical properties of colloidal plasmonic nanoparticles. Colloids Surf. A Physicochem. Eng. Asp. 2022, 640, 128521. [Google Scholar] [CrossRef]
- Ninakanti, R.; Dingenen, F.; Borah, R.; Peeters, H.; Verbruggen, S.W. Plasmonic Hybrid Nanostructures in Photocatalysis: Structures, Mechanisms, and Applications. Top. Curr. Chem. 2022, 380, 40. [Google Scholar] [CrossRef]
- Dingenen, F.; Blommaerts, N.; Van Hal, M.; Borah, R.; Arenas-Esteban, D.; Lenaerts, S.; Bals, S.; Verbruggen, S.W. Layer-by-Layer-Stabilized Plasmonic Gold-Silver Nanoparticles on TiO2: Towards Stable Solar Active Photocatalysts. Nanomaterials 2021, 11, 2624. [Google Scholar] [CrossRef] [PubMed]
- Peeters, H.; Keulemans, M.; Nuyts, G.; Vanmeert, F.; Li, C.; Minjauw, M.; Detavernier, C.; Bals, S.; Lenaerts, S.; Verbruggen, S.W. Plasmonic gold-embedded TiO2 thin films as photocatalytic self-cleaning coatings. Appl. Catal. B Environ. 2020, 267, 118654. [Google Scholar] [CrossRef]
- Borah, R.; Verbruggen, S.W. Silver–Gold Bimetallic Alloy versus Core–Shell Nanoparticles: Implications for Plasmonic Enhancement and Photothermal Applications. J. Phys. Chem. C 2020, 124, 12081–12094. [Google Scholar] [CrossRef]
- Darie, M.; Seftel, E.M.; Mertens, M.; Ciocarlan, R.G.; Cool, P.; Carja, G. Harvesting solar light on a tandem of Pt or Pt-Ag nanoparticles on layered double hydroxides photocatalysts for p-nitrophenol degradation in water. Appl. Clay Sci. 2019, 182, 105250. [Google Scholar] [CrossRef]
- Molina, P.M.; Meulendijks, N.; Xu, M.; Verheijen, M.A.; den Hartog, T.; Buskens, P.; Sastre, F. Low Temperature Sunlight-Powered Reduction of CO2 to CO Using a Plasmonic Au/TiO2 Nanocatalyst. ChemCatChem 2021, 13, 4507–4513. [Google Scholar] [CrossRef]
- Baffou, G.; Bordacchini, I.; Baldi, A.; Quidant, R. Simple experimental procedures to distinguish photothermal from hot-carrier processes in plasmonics. Light Sci. Appl. 2020, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Baffou, G.; Quidant, R. Nanoplasmonics for chemistry. Chem. Soc. Rev. 2014, 43, 3898–3907. [Google Scholar] [CrossRef]
- Asapu, R.; Claes, N.; Ciocarlan, R.-G.; Minjauw, M.; Detavernier, C.; Cool, P.; Bals, S.; Verbruggen, S.W. Electron Transfer and Near-Field Mechanisms in Plasmonic Gold-Nanoparticle-Modified TiO2 Photocatalytic Systems. ACS Appl. Nano Mater. 2019, 2, 4067–4074. [Google Scholar] [CrossRef]
- Baffou, G.; Cichos, F.; Quidant, R. Applications and challenges of thermoplasmonics. Nat. Mater. 2020, 19, 946–958. [Google Scholar] [CrossRef]
- Dubi, Y.; Un, I.W.; Sivan, Y. Thermal effects-an alternative mechanism for plasmon-assisted photocatalysis. Chem. Sci. 2020, 11, 5017–5027. [Google Scholar] [CrossRef]
- Kamarudheen, R.; Aalbers, G.J.W.; Hamans, R.F.; Kamp, L.P.J.; Baldi, A. Distinguishing among All Possible Activation Mechanisms of a Plasmon-Driven Chemical Reaction. ACS Energy Lett. 2020, 5, 2605–2613. [Google Scholar] [CrossRef]
- Un, I.-W.; Sivan, Y. Thermal effect in plasmon assisted photocatalyst: A parametric study. In Proceedings of the OSA Advanced Photonics Congress (AP) 2020 (IPR, NP, NOMA, Networks, PVLED, PSC, SPPCom, SOF), Washington, DC, USA, 13 July 2020; p. NoTh3C.3. [Google Scholar]
- Dubi, Y.; Sivan, Y. “Hot” electrons in metallic nanostructures—Non-thermal carriers or heating? Light Sci. Appl. 2019, 8, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahir, M.; Tahir, B.; Amin, N.A.S.; Zakaria, Z.Y. Photo-induced reduction of CO2 to CO with hydrogen over plasmonic Ag-NPs/TiO2 NWs core/shell hetero-junction under UV and visible light. J. CO2 Util. 2017, 18, 250–260. [Google Scholar] [CrossRef]
- Robatjazi, H.; Zhao, H.; Swearer, D.F.; Hogan, N.J.; Zhou, L.; Alabastri, A.; McClain, M.J.; Nordlander, P.; Halas, N.J. Plasmon-induced selective carbon dioxide conversion on earth-abundant aluminum-cuprous oxide antenna-reactor nanoparticles. Nat. Commun. 2017, 8, 27. [Google Scholar] [CrossRef]
- Upadhye, A.A.; Ro, I.; Zeng, X.; Kim, H.J.; Tejedor, I.; Anderson, M.A.; Dumesic, J.A.; Huber, G.W. Plasmon-enhanced reverse water gas shift reaction over oxide supported Au catalysts. Catal. Sci. Technol. 2015, 5, 2590–2601. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, T.; Wang, J.; Liu, H.; Dao, T.D.; Li, M.; Liu, G.; Meng, X.; Chang, K.; Shi, L.; et al. Surface-Plasmon-Enhanced Photodriven CO2 Reduction Catalyzed by Metal–Organic-Framework-Derived Iron Nanoparticles Encapsulated by Ultrathin Carbon Layers. Adv. Mater. 2016, 28, 3703–3710. [Google Scholar] [CrossRef]
- Wu, D.; Deng, K.; Hu, B.; Lu, Q.; Liu, G.; Hong, X. Plasmon-Assisted Photothermal Catalysis of Low-Pressure CO2 Hydrogenation to Methanol over Pd/ZnO Catalyst. ChemCatChem 2019, 11, 1598–1601. [Google Scholar] [CrossRef]
- Jia, J.; O’Brien, P.G.; He, L.; Qiao, Q.; Fei, T.; Reyes, L.M.; Burrow, T.E.; Dong, Y.; Liao, K.; Varela, M.; et al. Visible and Near-Infrared Photothermal Catalyzed Hydrogenation of Gaseous CO2 over Nanostructured Pd@Nb2O5. Adv. Sci. 2016, 3, 1600189. [Google Scholar] [CrossRef]
- Jia, J.; Wang, H.; Lu, Z.; O’Brien, P.G.; Ghoussoub, M.; Duchesne, P.; Zheng, Z.; Li, P.; Qiao, Q.; Wang, L.; et al. Photothermal Catalyst Engineering: Hydrogenation of Gaseous CO2 with High Activity and Tailored Selectivity. Adv. Sci. 2017, 4, 1700252. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Liu, M.; Yang, B.; Shu, W.; Shen, Q.; Liu, M.; Zhou, J. Enhanced Photocatalytic Performance toward CO2 Hydrogenation over Nanosized TiO2-Loaded Pd under UV Irradiation. J. Phys. Chem. C 2017, 121, 2923–2932. [Google Scholar] [CrossRef]
- Ge, H.; Kuwahara, Y.; Kusu, K.; Yamashita, H. Plasmon-induced catalytic CO2 hydrogenation by a nano-sheet Pt/HxMoO3−y hybrid with abundant surface oxygen vacancies. J. Mater. Chem. A 2021, 9, 13898–13907. [Google Scholar] [CrossRef]
- Blommaerts, N.; Hoeven, N.; Esteban, D.A.; Campos, R.; Mertens, M.; Borah, R.; Glisenti, A.; De Wael, K.; Bals, S.; Lenaerts, S.; et al. Tuning the turnover frequency and selectivity of photocatalytic CO2 reduction to CO and methane using platinum and palladium nanoparticles on Ti-Beta zeolites. Chem. Eng. J. 2021, 410, 128234. [Google Scholar] [CrossRef]
- Ro, I.; Carrasquillo-Flores, R.; Dumesic, J.A.; Huber, G.W. Intrinsic kinetics of plasmon-enhanced reverse water gas shift on Au and Au–Mo interfacial sites supported on silica. Appl. Catal. A Gen. 2016, 521, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, X.; Zhang, D.; Su, N.Q.; Yang, W.; Everitt, H.O.; Liu, J. Product selectivity in plasmonic photocatalysis for carbon dioxide hydrogenation. Nat. Commun. 2017, 8, 14542. [Google Scholar] [CrossRef] [Green Version]
- Tahir, B.; Tahir, M.; Amin, N.A.S. Photocatalytic CO2 conversion over Au/TiO2 nanostructures for dynamic production of clean fuels in a monolith photoreactor. Clean Technol. Environ. Policy 2016, 18, 2147–2160. [Google Scholar] [CrossRef]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. The Formation of Colloidal Gold. J. Phys. Chem. 1953, 57, 670–673. [Google Scholar] [CrossRef]
- Mohamed, I.M.A.; Dao, V.-D.; Yasin, A.S.; Barakat, N.A.M.; Choi, H.-S. Design of an efficient photoanode for dye-sensitized solar cells using electrospun one-dimensional GO/N-doped nanocomposite SnO2/TiO2. Applied Surface Science 2017, 400, 355–364. [Google Scholar] [CrossRef]
- Gates-Rector, S.; Blanton, T. The Powder Diffraction File: A quality materials characterization database. Powder Diffraction 2019, 34, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Francesca, S.; Teresa, F.M.; Teresa, P.; Nicolas, G. Mesoporous TiO2 Thin Films: State of the Art. In Titanium Dioxide; Dongfang, Y., Ed.; IntechOpen: Rijeka, Croatia, 2018; pp. 57–80. [Google Scholar]
- Ren, X.; Song, Y.; Liu, A.; Zhang, J.; Yang, P.; Zhang, J.; An, M. Experimental and theoretical studies of DMH as a complexing agent for a cyanide-free gold electroplating electrolyte. RSC Adv. 2015, 5, 64997–65004. [Google Scholar] [CrossRef]
- Xu, M.; den Hartog, T.; Cheng, L.; Wolfs, M.; Habets, R.; Rohlfs, J.; van den Ham, J.; Meulendijks, N.; Sastre, F.; Buskens, P. Using Fiber Bragg Grating Sensors to Quantify Temperature Non-Uniformities in Plasmonic Catalyst Beds under Illumination. ChemPhotoChem 2022, 6, e202100289. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Everitt, H.O.; Liu, J. Light-Induced Thermal Gradients in Ruthenium Catalysts Significantly Enhance Ammonia Production. Nano Lett. 2019, 19, 1706–1711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Reish, M.E.; Zhang, D.; Su, N.Q.; Gutiérrez, Y.; Moreno, F.; Yang, W.; Everitt, H.O.; Liu, J. Plasmon-Enhanced Catalysis: Distinguishing Thermal and Nonthermal Effects. Nano Lett. 2018, 18, 1714–1723. [Google Scholar] [CrossRef]
- Mutschler, R.; Moioli, E.; Zhao, K.; Lombardo, L.; Oveisi, E.; Porta, A.; Falbo, L.; Visconti, C.G.; Lietti, L.; Züttel, A. Imaging Catalysis: Operando Investigation of the CO2 Hydrogenation Reaction Dynamics by Means of Infrared Thermography. ACS Catal. 2020, 10, 1721–1730. [Google Scholar] [CrossRef]
- Sivan, Y.; Baraban, J.H.; Dubi, Y. Experimental practices required to isolate thermal effects in plasmonic photo-catalysis: Lessons from recent experiments. OSA Contin. 2020, 3, 483–497. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volders, J.; Elen, K.; Raes, A.; Ninakanti, R.; Kelchtermans, A.-S.; Sastre, F.; Hardy, A.; Cool, P.; Verbruggen, S.W.; Buskens, P.; et al. Sunlight-Powered Reverse Water Gas Shift Reaction Catalysed by Plasmonic Au/TiO2 Nanocatalysts: Effects of Au Particle Size on the Activity and Selectivity. Nanomaterials 2022, 12, 4153. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12234153
Volders J, Elen K, Raes A, Ninakanti R, Kelchtermans A-S, Sastre F, Hardy A, Cool P, Verbruggen SW, Buskens P, et al. Sunlight-Powered Reverse Water Gas Shift Reaction Catalysed by Plasmonic Au/TiO2 Nanocatalysts: Effects of Au Particle Size on the Activity and Selectivity. Nanomaterials. 2022; 12(23):4153. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12234153
Chicago/Turabian StyleVolders, Jordi, Ken Elen, Arno Raes, Rajeshreddy Ninakanti, An-Sofie Kelchtermans, Francesc Sastre, An Hardy, Pegie Cool, Sammy W. Verbruggen, Pascal Buskens, and et al. 2022. "Sunlight-Powered Reverse Water Gas Shift Reaction Catalysed by Plasmonic Au/TiO2 Nanocatalysts: Effects of Au Particle Size on the Activity and Selectivity" Nanomaterials 12, no. 23: 4153. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12234153