
Molecular Dynamics Simulation of the Thermal Behavior of Hydroxyapatite

 ,
,
Abstract
:
1. Introduction
2. Computational Details, Main Models, and Methods
2.1. Main Methods and Used Software
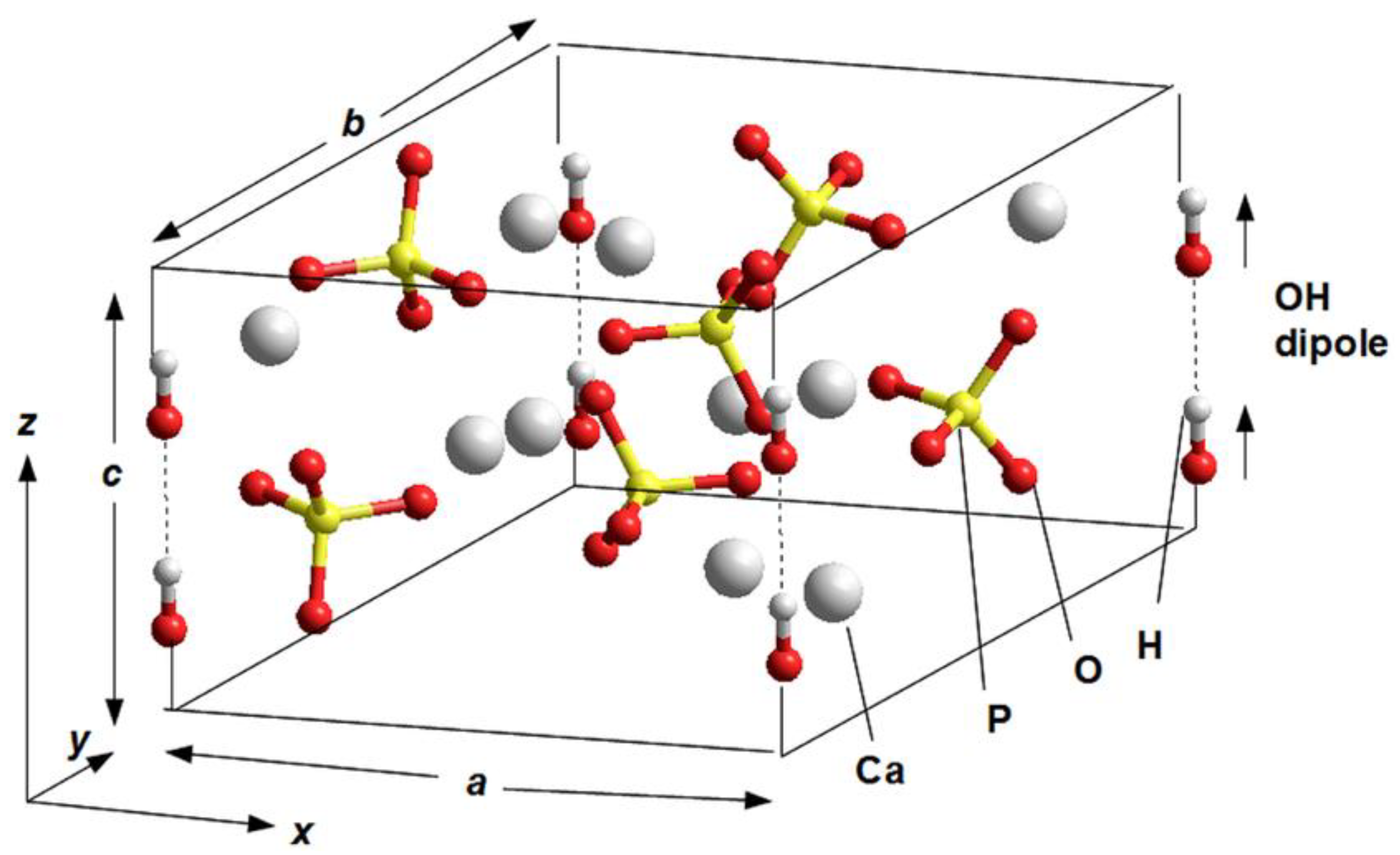
2.2. Initial Structural Data
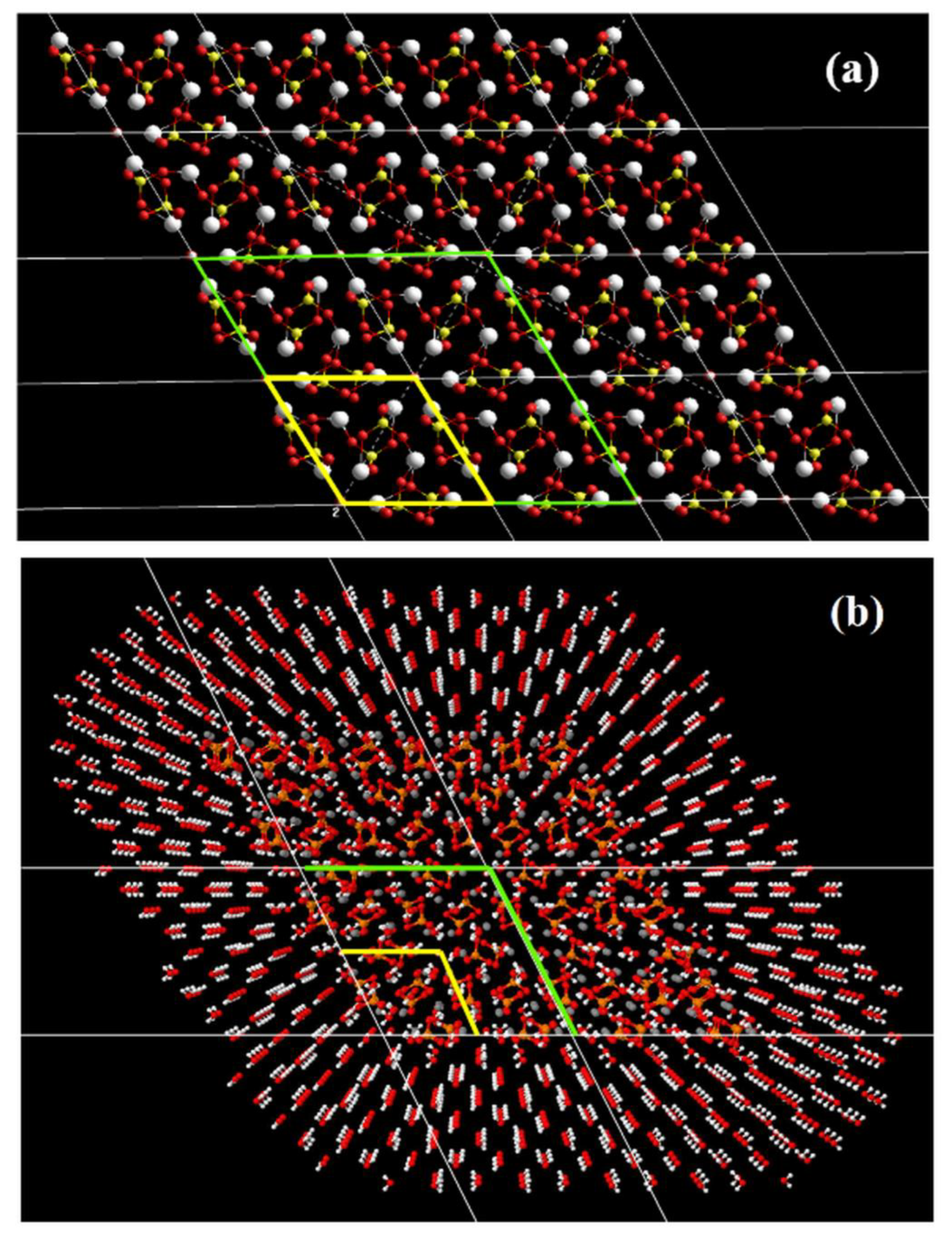
2.3. Simulation Procedure
2.4. Algorithm for Simulation Procedure Analysis
2.5. Evaluation of the Energy of the HAP System upon Removal of OH Groups
2.6. Evaluation of the Dynamics of HAP Behavior at Different Temperatures
3. Results and Discussion
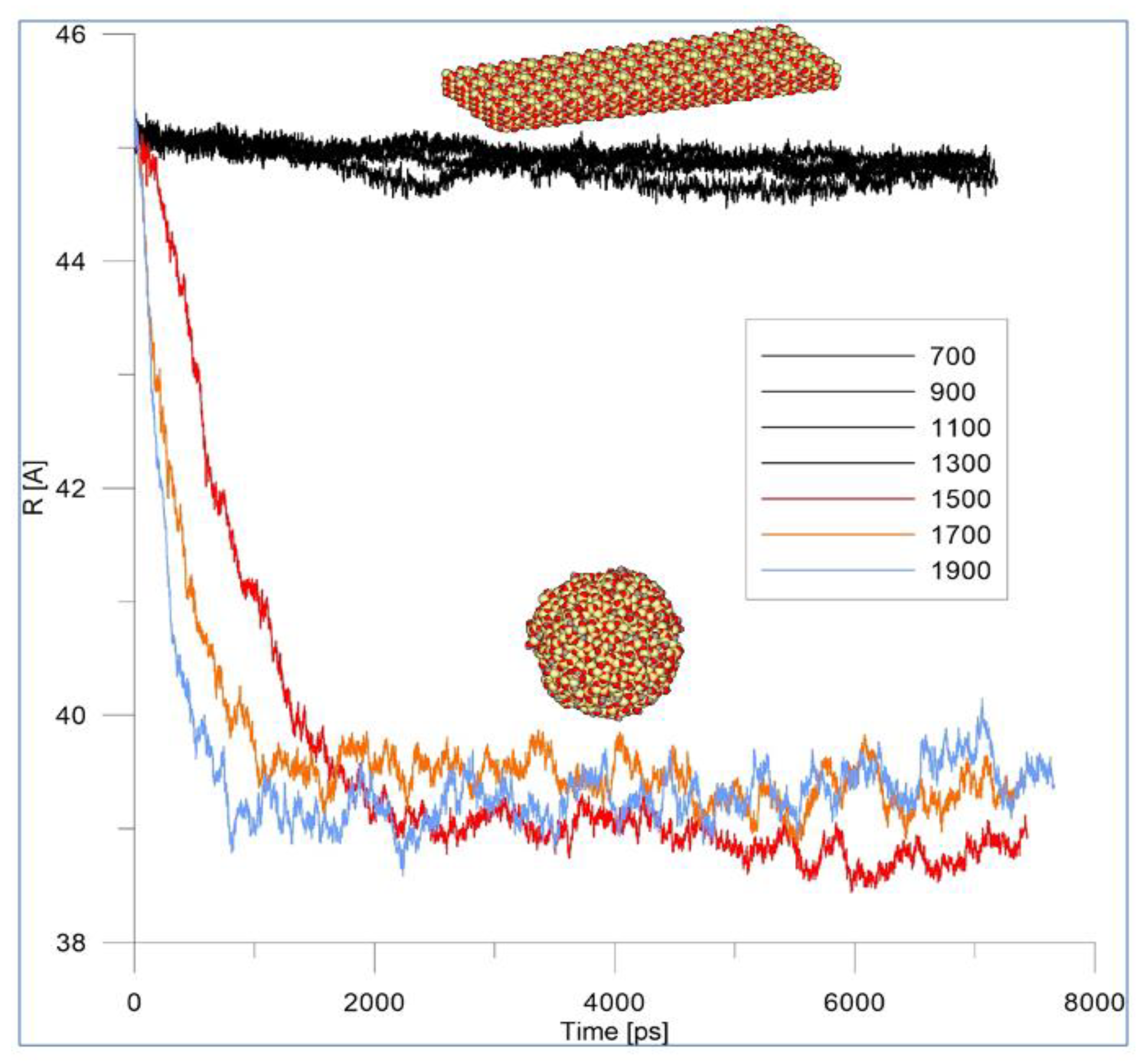
3.1. Dynamics of HAP Model Behavior and Melting at Different Temperatures
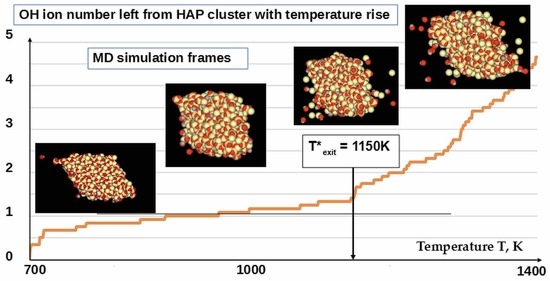
3.2. Detachment of OH Ions at Heating
3.3. Changes of the Energy of the HAP System upon Removal of OH Groups

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
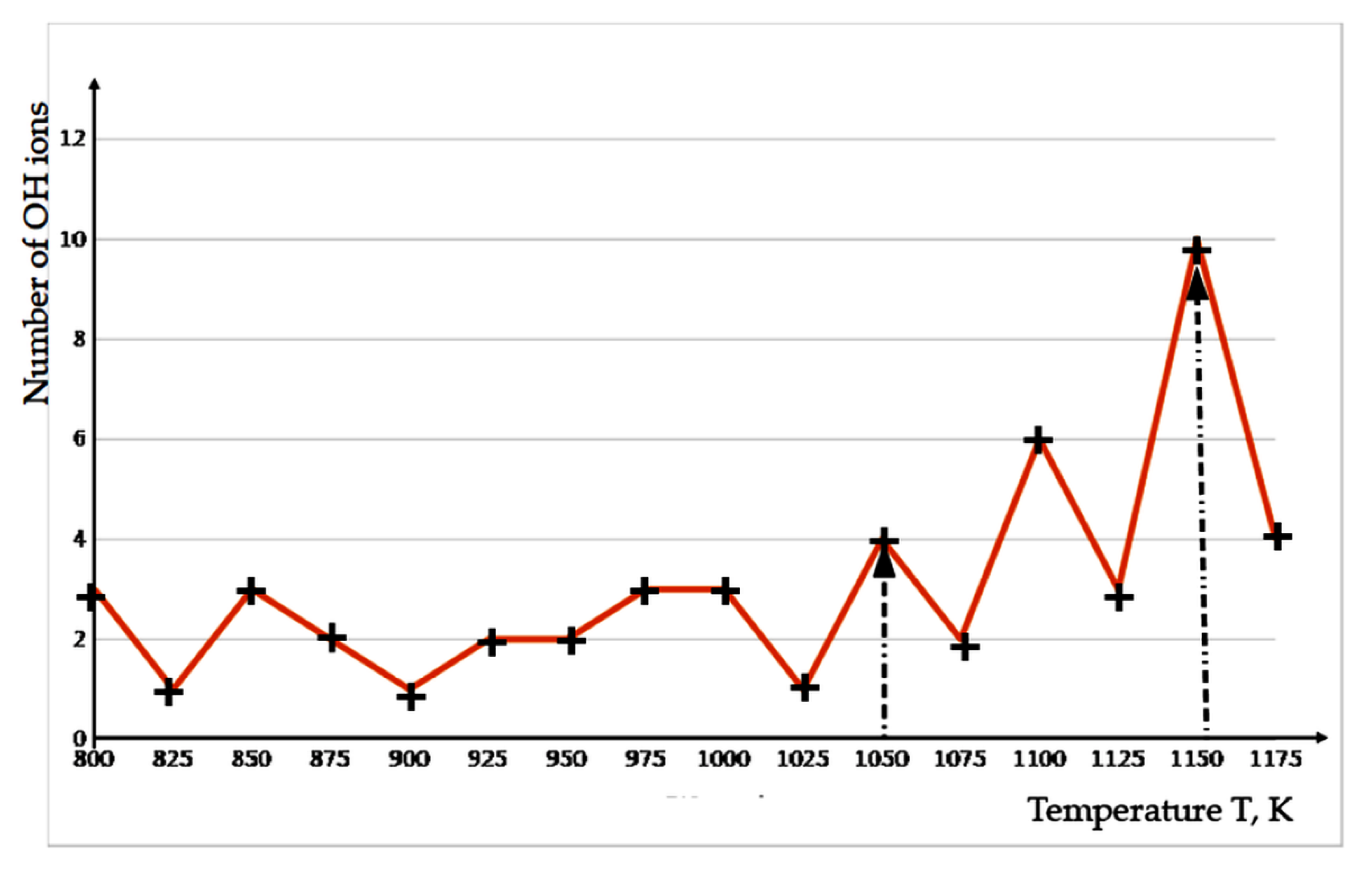{kind=link}
{kind=link}
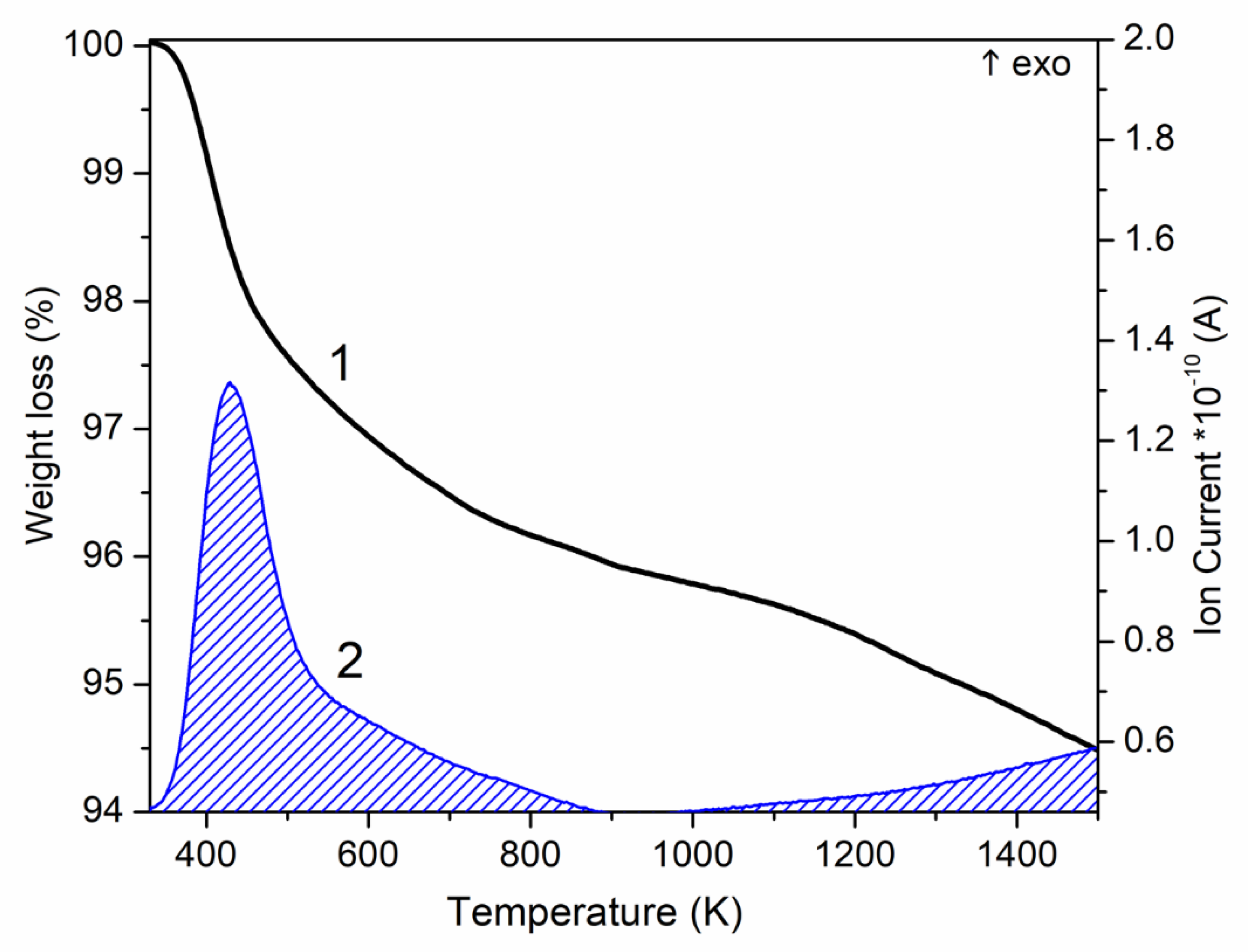{kind=link}
{kind=link}
{kind=link}
| Model | Total Energy Change |ΔE|, kcal/mol | Energy Change per 1 Unit Cell |ΔE1|, kcal/mol | Energy Change per 1 Unit Cell |ΔE1|, eV | Corresponding Temperature T=|ΔE1|/kB, K | |
|---|---|---|---|---|---|
| 1 | HAP32—1_OH (OH from one OH-channel) | 55.21 ± 5.37 | 1.725 ± 0.167 | 0.075 ± 0.007 | 870 ± 84 |
| 2 | HAP32—2_OH (from one OH-channel) | 69.33 ± 6.73 | 2.167 ± 0.210 | 0.094 ± 0.009 | 1090 ± 106 |
| 3 | HAP32—2_OH (from two OH-channel) | 70.62 ± 6.85 | 2.207 ± 0.214 | 0.096 ± 0.009 | 1114 ± 108 |
| 4 | HAP32—3_OH (from one OH-channel) | 101.13 ± 9.81 | 3.160 ± 0.307 | 0.137 ± 0.013 | 1590 ± 154 |
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ratner, B.D.; Hoffman, A.S.; Schoen, F.J.; Lemons, J.E. Biomaterials Science, 3rd ed.; Academic Press: Oxford, UK, 2013. [Google Scholar]
- Ducheyne, P.; Healy, K.; Hutmacher, D.E.; Grainger, D.W.; Kirkpatrick, C.J. (Eds.) Comprehensive Biomaterials II, 2nd ed.; Seven-Volume Set; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Dorozhkin, S.V. Calcium Orthophosphate (CaPO4)-Based Bioceramics: Preparation, Properties, and Applications. Coatings 2022, 12, 1380. [Google Scholar] [CrossRef]
- Bystrov, V.; Bystrova, A.; Dekhtyar, Y.; Khlusov, I.A.; Pichugin, V.; Prosolov, K.; Sharkeev, Y. Electrical functionalization and fabrication of nanostructured hydroxyapatite coatings. In Bioceramics and Biocomposites: From Research to Clinical Practice; Jiulian, A., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2019; pp. 149–190. [Google Scholar]
- Leon, B.; Janson, J.A. Thin Calcium Phosphate Coatings for Medical Implants; Springer: Berlin, Germany, 2009. [Google Scholar]
- Baltacis, K.; Bystrov, V.; Bystrova, A.; Dekhtyar, Y.; Freivalds, T.; Raines, J.; Rozenberga, K.; Sorokins, H.; Zeidaks, M. Physical fundamentals of biomaterials surface electrical functionalization. Materials 2020, 13, 4575. [Google Scholar] [CrossRef] [PubMed]
- Bystrov, V.; Paramonova, E.; Avakyan, L.; Coutinho, J.; Bulina, N. Simulation and Computer Study of Structures and Physical Properties of Hydroxyapatite with Various Defects. Nanomaterials 2021, 11, 2752. [Google Scholar] [CrossRef] [PubMed]
- Bystrov, V.S.; Coutinho, J.; Bystrova, A.V.; Dekhtyar, Y.D.; Pullar, R.C.; Poronin, A.; Palcevskis, E.; Dindune, A.; Alkan, B.; Durucan, C. Computational study of the hydroxyapatite structures, properties and defects. J. Phys. D Appl. Phys. 2015, 48, 195302. [Google Scholar] [CrossRef]
- Bystrov, V.S.; Piccirillo, C.; Tobaldi, D.M.; Castro, P.M.L.; Coutinho, J.; Kopyl, S.; Pullar, R.C. Oxygen vacancies, the optical band gap (Eg) and photocatalysis of hydroxyapatite: Comparing modelling with measured data. Appl. Catal. B Environ. 2016, 196, 100–107. [Google Scholar] [CrossRef]
- Figueroa-Rosales, E.X.; Martínez-Juárez, J.; García-Díaz, E.; Hernández-Cruz, D.; Sabinas-Hernández, S.A.; Robles-Águila, M.J. Photoluminescent Properties of Hydroxyapatite and Hydroxyapatite/Multi-Walled Carbon Nanotube Composites. Crystals 2021, 11, 832. [Google Scholar] [CrossRef]
- Avakyan, L.A.; Paramonova, E.V.; Coutinho, J.; Öberg, S.; Bystrov, V.S.; Bugaev, L.A. Optoelectronics and defect levels in hydroxyapatite by first-principles. J. Chem. Phys. 2018, 148, 154706. [Google Scholar] [CrossRef] [Green Version]
- Bystrov, V.S.; Avakyan, L.A.; Paramonova, E.V.; Coutinho, J. Sub-Band Gap Absorption Mechanisms Involving Oxygen Vacancies in Hydroxyapatite. J. Chem. Phys. C 2019, 123, 4856–4865. [Google Scholar] [CrossRef] [Green Version]
- Bulina, N.V.; Makarova, S.V.; Baev, S.G.; Matvienko, A.A.; Gerasimov, K.B.; Logutenko, O.A.; Bystrov, V.S. A Study of Thermal Stability of Hydroxyapatite. Minerals 2021, 11, 1310. [Google Scholar] [CrossRef]
- Eremina, N.V.; Makarova, S.V.; Isaev, D.D.; Bulina, N.V. Soft mechanochemical synthesis and thermal stability of hydroxyapatites with different types of substitution. Chim. Techno Acta 2022, 9, 20229305. [Google Scholar] [CrossRef]
- Tõnsuaadu, K.; Gross, K.A.; Pluduma, L.; Veiderma, M. A review on the thermal stability of calcium apatites. J. Therm. Anal. Calorim. 2012, 110, 647–659. [Google Scholar] [CrossRef]
- Elliott, J. Structure and Chemistry of the Apatites and Other Calcium Orthophosphates, Studies in Inorganic Chemistry; Elsevier Science: Amsterdam, The Ntherlands, 2013; p. 404. [Google Scholar]
- Hughes, J.M.; Cameron, M.; Crowley, K.D. Structural variations in natural F, OH, and Cl apatites. Am. Mineral. 1989, 74, 870–876. Available online: http://rruff.geo.arizona.edu/AMS/result.php (accessed on 1 June 2022).
- Kay, M.I.; Young, R.A.; Posner, A.S. Crystal structure of hydroxyapatite. Nature 1964, 204, 1050. [Google Scholar] [CrossRef]
- Marković, S.; Veselinović, L.; Lukić, M.J.; Karanović, L.; Bračko, I.; Ignjatović, N.; Uskoković, D. Synthetical bone-like and biological hydroxyapatites: A comparative study of crystal structure and morphology. Biomed. Mater. 2011, 6, 045005. [Google Scholar] [CrossRef] [PubMed]
- Hitmi, N.; LaCabanne, C.; Young, R.A. OH- dipole reorientability in hydroxyapatites: Effect of tunnel size. J. Phys. Chem. Solids 1986, 47, 533–546. [Google Scholar] [CrossRef]
- Bystrov, V.S.; Bystrova, N.K.; Paramonova, E.V.; Dekhtyar, Y.D. Interaction of charged hydroxyapatite and living cells. I. Hydroxyapatite polarization properties. Math. Biol. Bioinform. 2009, 4, 7–11. Available online: http://www.matbio.org/downloads_en/Bystrov_en2009(4_7).pdf (accessed on 1 October 2022).
- Nakamura, S.; Takeda, H.; Yamashita, K. Proton transport polarization and depolarization of hydroxyapatite ceramics. J. Appl. Phys. 2001, 89, 5386. [Google Scholar] [CrossRef]
- Horiuchi, N.; Nakamura, M.; Nagai, A.; Katayama, K.; Yamashita, K. Proton conduction related electrical dipole and space charge polarization in hydroxyapatite. J. Appl. Phys. 2012, 112, 074901. [Google Scholar] [CrossRef]
- Tofail, S.A.M.; Haverty, D.; Stanton, K.T.; McMonagle, J.B. Structural order and dielectric behaviour of hydroxyapatite. Ferroelectrics 2005, 319, 117–123. [Google Scholar] [CrossRef]
- Matsunaga, K.; Kuwabara, A. First-principles study of vacancy formation in hydroxyapatite. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 75, 014102. [Google Scholar] [CrossRef] [Green Version]
- Aronov, D.; Chaikina, M.; Haddad, J.; Karlov, A.; Mezinskis, G.; Oster, L.; Pavlovska, I.; Rosenman, G. Electronic states spectroscopy of Hydroxyapatite ceramics. J. Mater. Sci. Mater. Med. 2007, 18, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H. A high active type of hydroxyapatite for photocatalytic decomposition of dimethyl sulfide under UV irradiation. J. Mol. Catal. A Chem. 2004, 207, 149–153. [Google Scholar] [CrossRef]
- Nishikawa, H. Photo-induced catalytic activity of hydroxyapatite based on photo-excitation. Phosphorus Res. Bull. 2007, 21, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Silin, A.R.; Trukhin, A.I. Point Defects and Elementary Excitations in Crystalline and Glassy SiO2; Zinatne: Riga, Latvia, 1985. (In Russian) [Google Scholar]
- Avakyan, L.; Paramonova, E.; Bystrov, V.; Coutinho, J.; Gomes, S.; Renaudin, G. Iron in Hydroxyapatite: Interstitial or Substitution Sites? Nanomaterials 2021, 11, 2978. [Google Scholar] [CrossRef] [PubMed]
- Bystrov, V.S.; Paramonova, E.V.; Bystrova, A.V.; Avakyan, L.A.; Bulina, N.V. Structural and physical properties of Sr/Ca and Mg/Ca substituted hydroxyapatite: Modeling and experiments. Ferroelectrics 2022, 590, 41–48. [Google Scholar] [CrossRef]
- Bulina, N.V.; Makarova, S.V.; Prosanov, I.Y.; Vinokurova, O.B.; Lyakhov, N.Z. Structure and thermal stability of fluorhydroxyapatite and fluorapatite obtained by mechanochemical method. J. Solid State Chem. 2020, 282, 121076. [Google Scholar] [CrossRef]
- Bulina, N.V.; Chaikina, M.V.; Prosanov, I.Y. Mechanochemical Synthesis of Sr-Substituted Hydroxyapatite. Inorg. Mater. 2018, 54, 820–825. [Google Scholar] [CrossRef]
- Šupova, M. Substituted hydroxyapatites for biomedical applications: A review. Ceram. Int. 2015, 41, 9203–9231. [Google Scholar] [CrossRef]
- Capuccini, C.; Torricelli, P.; Boanini, E.; Gazzano, M.; Giardino, R.; Bigi, A. Interaction of Sr-doped hydroxyapatite nanocrystals with osteoclast and osteoblast-like cells. J. Biomed. Mater. Res. Part A 2009, 89, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Bystrov, V.S.; Coutinho, J.; Avakyan, L.A.; Bystrova, A.V.; Paramonova, E.V. Piezoelectric, ferroelectric, and optoelectronic phenomena in hydroxyapatite with various defect levels. Ferroelectrics 2020, 550, 45–55. [Google Scholar] [CrossRef]
- Calderin, L.; Stott, M.J.; Rubio, A. Electronic and crystallographic structure of apatites. Phys. Rev. B Condens. Matter Mater. Phys. 2003, 67, 134106. [Google Scholar] [CrossRef] [Green Version]
- Rulis, P.; Yao, H.; Ouyang, L.; Ching, W.Y. Electronic structure, bonding, charge distribution, and X-ray absorption spectra of the (001) surfaces of fluorapatite and hydroxyapatite from first principles. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 76, 245410. [Google Scholar] [CrossRef]
- Haverty, D.; Tofail, S.A.M.; Stanton, K.T.; McMonagle, J.B. Structure and stability of hydroxyapatite: Density functional calculation and Rietveld analysis. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 71, 094103. [Google Scholar] [CrossRef]
- Slepko, A.; Demkov, A.A. First-principles study of the biomineral hydroxyapatite. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 84, 134108. [Google Scholar] [CrossRef] [Green Version]
- Corno, M.; Busco, C.; Civalleri, B.; Ugliengo, P. Periodic ab initio study of structural and vibrational features of hexagonal hydroxyapatite Ca10(PO4)6(OH)2. Phys. Chem. Chem. Phys. 2006, 8, 2464. [Google Scholar] [CrossRef]
- Britney, P.R.; Jones, R. LDA Calculations Using a Basis of Gaussian Orbitals. Phys. Status Solidi B Basic Res. 2000, 217, 131–171. [Google Scholar]
- Briddon, P.R.; Rayson, M.J. Accurate Kohn–Sham DFT with the Speed of Tight Binding: Current Techniques and Future Directions in Materials Modeling. Phys. Status Solidi B 2011, 248, 1309–1318. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. A new mixing of Hartree-Fock and local density functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- AIMPRO, 2010. Available online: http://aimpro.ncl.ac.uk/ (accessed on 22 February 2016).
- VASP (Vienna Ab Initio Simulation Package). Available online: https://www.vasp.at/ (accessed on 1 July 2019).
- Quantum ESPRESSO. Available online: https://www.quantum-espresso.org/ (accessed on 1 July 2022).
- Hyper Chem: Tools for Molecular Modeling (Release 8); Hypercube, Inc.: Gainnesville, FL, USA, 2011.
- Stewart, J.J.P. Computational Chemistry. MOPAC2016, Colorado Springs, USA, 2016. Available online: http://openmopac.net/MOPAC2016.html (accessed on 30 July 2022).
- Kamberaj, H. Molecular Dynamics Simulations in Statistical Physics: Theory and Applications; Springer Nature: Cham, Switzerland, 2020. [Google Scholar]
- Brooks, C.L., III; Case, D.A.; Plimpton, S.; Roux, B.; Van Der Spoel, D.; Tajkhorshid, E. Classical molecular dynamics. J. Chem. Phys. 2021, 154, 100401. [Google Scholar] [CrossRef]
- Grigoriev, F.V.; Sulimov, V.B.; Tikhonravov, A.V. Molecular Dynamics Simulation of Laser Induced Heating of Silicon Dioxide Thin Films. Nanomaterials 2021, 11, 2986. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. Available online: http://0-www-sciencedirect-com.brum.beds.ac.uk/science/article/pii/S2352711015000059 (accessed on 17 September 2022). [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [Green Version]
- LLindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Systematic Validation of Protein Force Fields against Experimental Data. PLoS ONE 2012, 7, e32131. [Google Scholar] [CrossRef] [Green Version]
- Voevodin, V.L.; Antonov, A.; Nikitenko, D.; Shvets, P.; Sobolev, S.; Sidorov, I.; Stefanov, K.; Voevodin, V.; Zhumatiy, S. Supercomputer Lomonosov-2: Large Scale, Deep Monitoring and Fine Analytics for the User Community. Supercomput. Front. Innov. 2019, 6, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Lemak, A.S.; Balabaev, N.K. A Comparison between Collisional Dynamics and Brownian Dynamics. Mol. Simul. 1995, 15, 223–231. [Google Scholar] [CrossRef]
- Lemak, A.S.; Balabaev, N.K. Molecular Dynamics Simulation of a Polymer Chain in Solution by Collisional Dynamics Method. J. Comput. Chem. 1996, 17, 1685–1695. [Google Scholar] [CrossRef]
- Balabaev, N.K.; Lemak, A.S. Molecular Dynamics of a Linear Polymer in a Hydrodynamic Flow. Russ. J. Phys. Chem. A 1995, 69, 24–32. [Google Scholar]
- Likhachev, I.V.; Balabaev, N.K. Parallelism of different levels in the program of molecular dynamics simulation PUMA-CUDA. In Proceedings of the International Conference “Mathematical Biology and Bioinformatics”, Pushchino, Russia, 14–19 October 2018; Lakhno, V.D., Ed.; IMPB RAS: Pushchino, Russia, 2018; Volume 7, p. e44. [Google Scholar] [CrossRef]
- Available online: https://www.kiam.ru/MVS/resourses/ (accessed on 1 October 2022).
- Likhachev, I.V.; Balabaev, N.K.; Galzitskaya, O.V. Available Instruments for Analyzing Molecular Dynamics Trajectories. Open Biochem. J. 2016, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Likhachev, I.V.; Balabaev, N.K. Trajectory Analyzer of Molecular Dynamics. Math. Biol. Bioinform. 2007, 2, 120–129. [Google Scholar] [CrossRef]
- Likhachev, I.V.; Balabaev, N.K. Construction of Extended Dynamical Contact Maps by Molecular-Dynamics Simulation Data. Math. Biol. Bioinform. 2009, 4, 36–45. [Google Scholar] [CrossRef]
- Lin, T.-J.; Heinz, H. Accurate Force Field Parameters and pH Resolved Surface Models for Hydroxyapatite to Understand Structure, Mechanics, Hydration, and Biological Interfaces. J. Phys. Chem. C 2016, 120, 4975–4992. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.M.; Cieplak, P.; Kollman, P. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J. Comput. Chem. 1999, 21, 1049. [Google Scholar] [CrossRef]
- Mahoney, M.W.; Jorgensen, W.L. A Five-Site Model for Liquid Water and the Reproduction of the Density Anomaly by Rigid, Nonpolarizable Potential Functions. J. Chem. Phys. 2000, 112, 8910–8922. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Peka, M.; Lennart, N. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 86th ed.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]









| Calculation Using Method Reported in [11] | Experimental Data [76] | |||
|---|---|---|---|---|
| Compound | ΔfH, eV/(PO4)6 | |ΔE|, eV/(PO4)6 | ΔfH, eV/(PO4)6 | |ΔE|, eV/(PO4)6 |
| HAP | −128.49 | 0 | −138.88 | 0 |
| γ-TCP | −117.54 | 10.95 | −128.48 | 10.40 |
| β-TCP | −117.38 | 11.11 | ||
| HAP-1OH (without 1OH) | (−122.99) * | (5.0–5.5) * | ||
| HAP-2OH (without 2OH) | −117.68 | 10.81 | ||
| OAP | −123.16 | 5.33 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Likhachev, I.; Balabaev, N.; Bystrov, V.; Paramonova, E.; Avakyan, L.; Bulina, N. Molecular Dynamics Simulation of the Thermal Behavior of Hydroxyapatite. Nanomaterials 2022, 12, 4244. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12234244
Likhachev I, Balabaev N, Bystrov V, Paramonova E, Avakyan L, Bulina N. Molecular Dynamics Simulation of the Thermal Behavior of Hydroxyapatite. Nanomaterials. 2022; 12(23):4244. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12234244
Chicago/Turabian StyleLikhachev, Ilya, Nikolay Balabaev, Vladimir Bystrov, Ekaterina Paramonova, Leon Avakyan, and Natalia Bulina. 2022. "Molecular Dynamics Simulation of the Thermal Behavior of Hydroxyapatite" Nanomaterials 12, no. 23: 4244. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12234244