
Hydrogenated Amorphous Titania with Engineered Surface Oxygen Vacancy for Efficient Formaldehyde and Dye Removals under Visible-Light Irradiation


Abstract
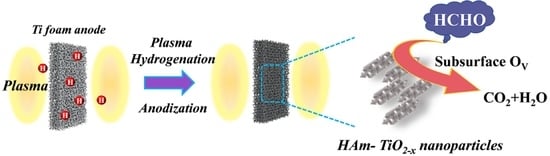
:
1. Introduction
2. Experimental Section
2.1. Reagents and Materials
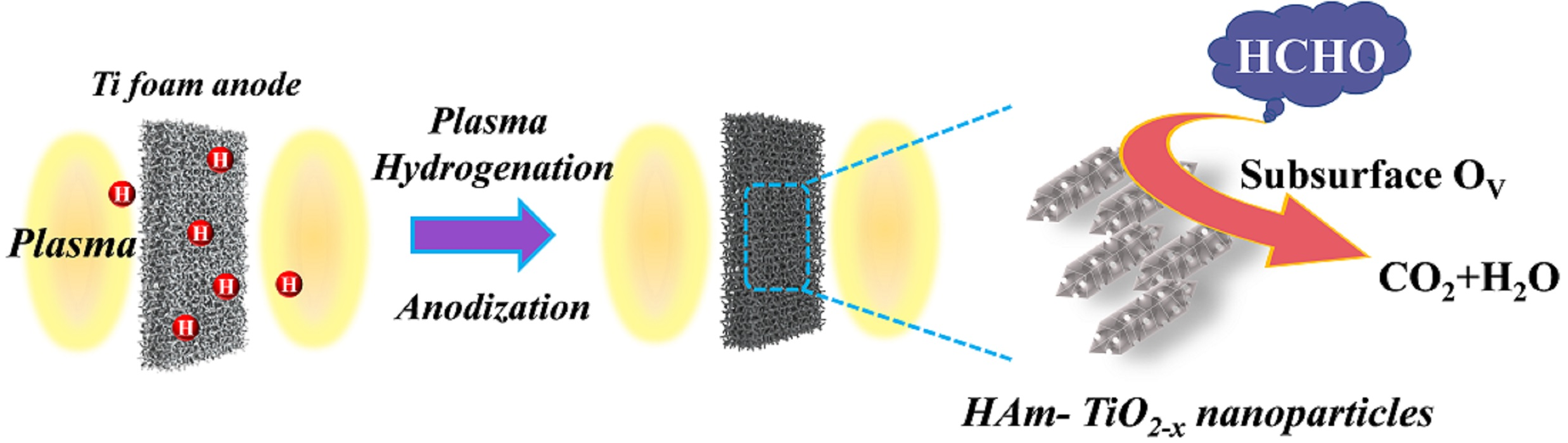
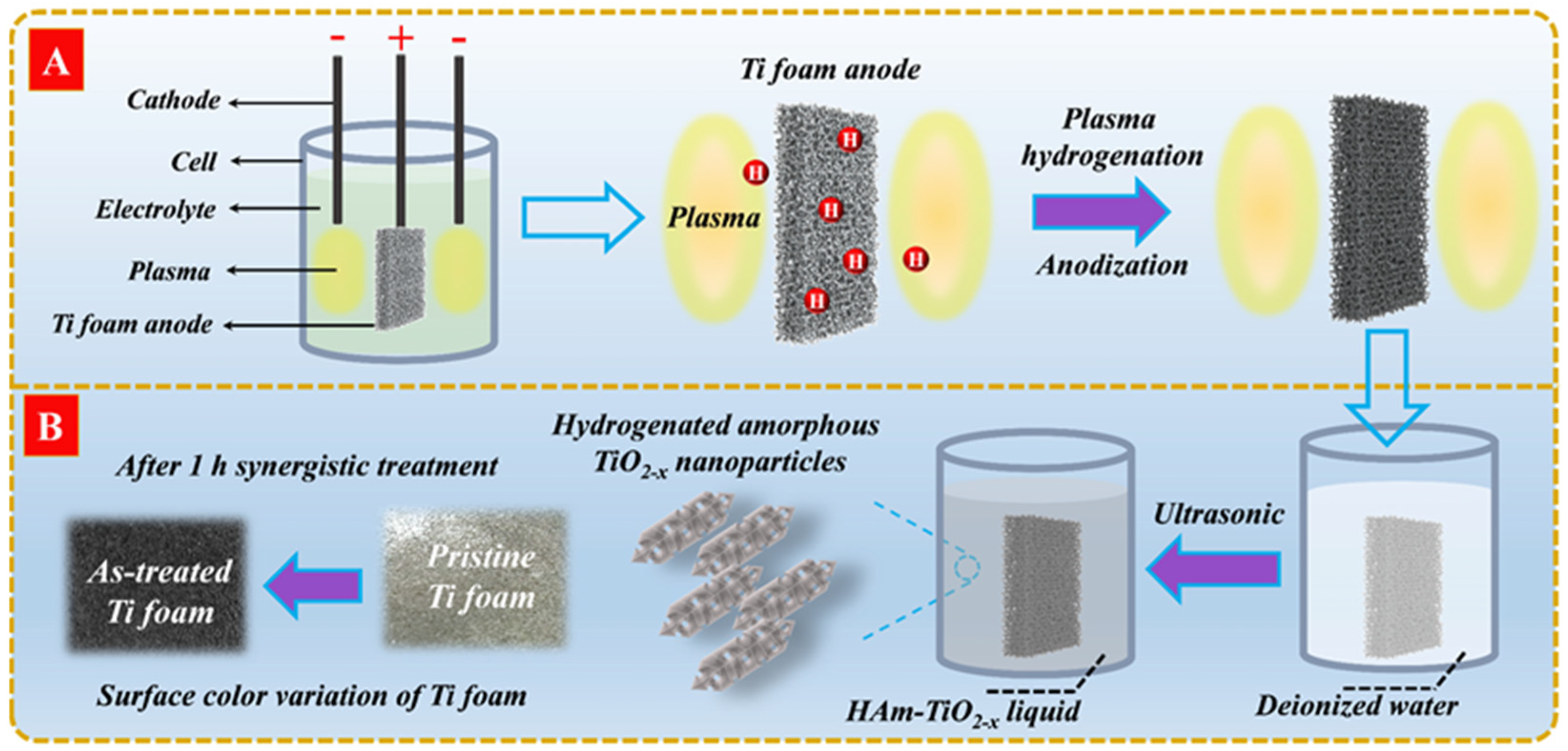
2.2. Preparation of HAm-TiO2−x
2.3. Preparation of HAm-TiO2−x-Coated Glass
2.4. Characterization
2.5. Photodegradation Experiments of HA-TiO2−x
2.5.1. The Dyes Photodegradation of HAm-TiO2−x Nanoparticles and Their Coatings
2.5.2. Formaldehyde (HCHO) Photodecomposition Test over HAm-TiO2−x
2.6. Theoretical Calculation Methods
3. Results and Discussion
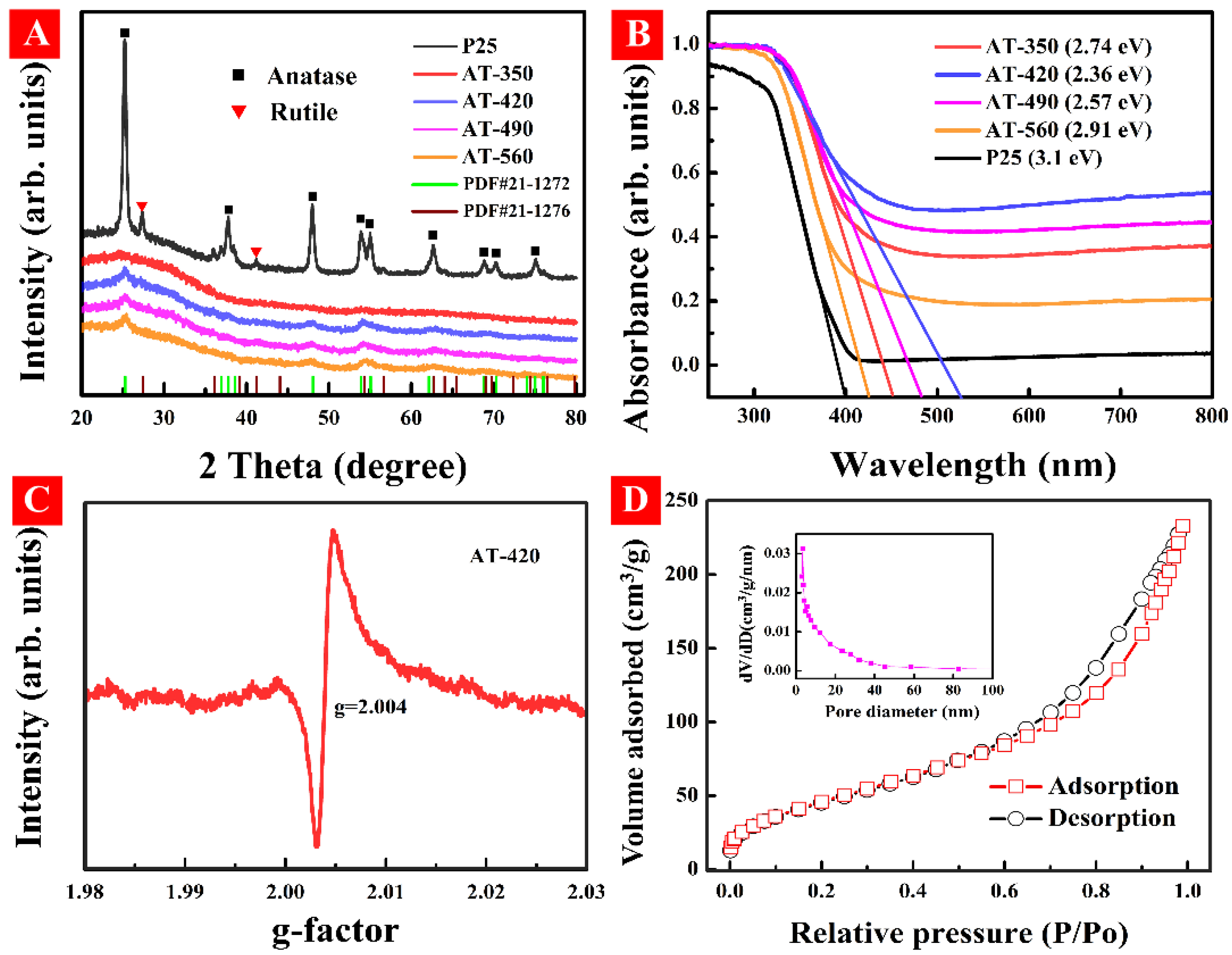
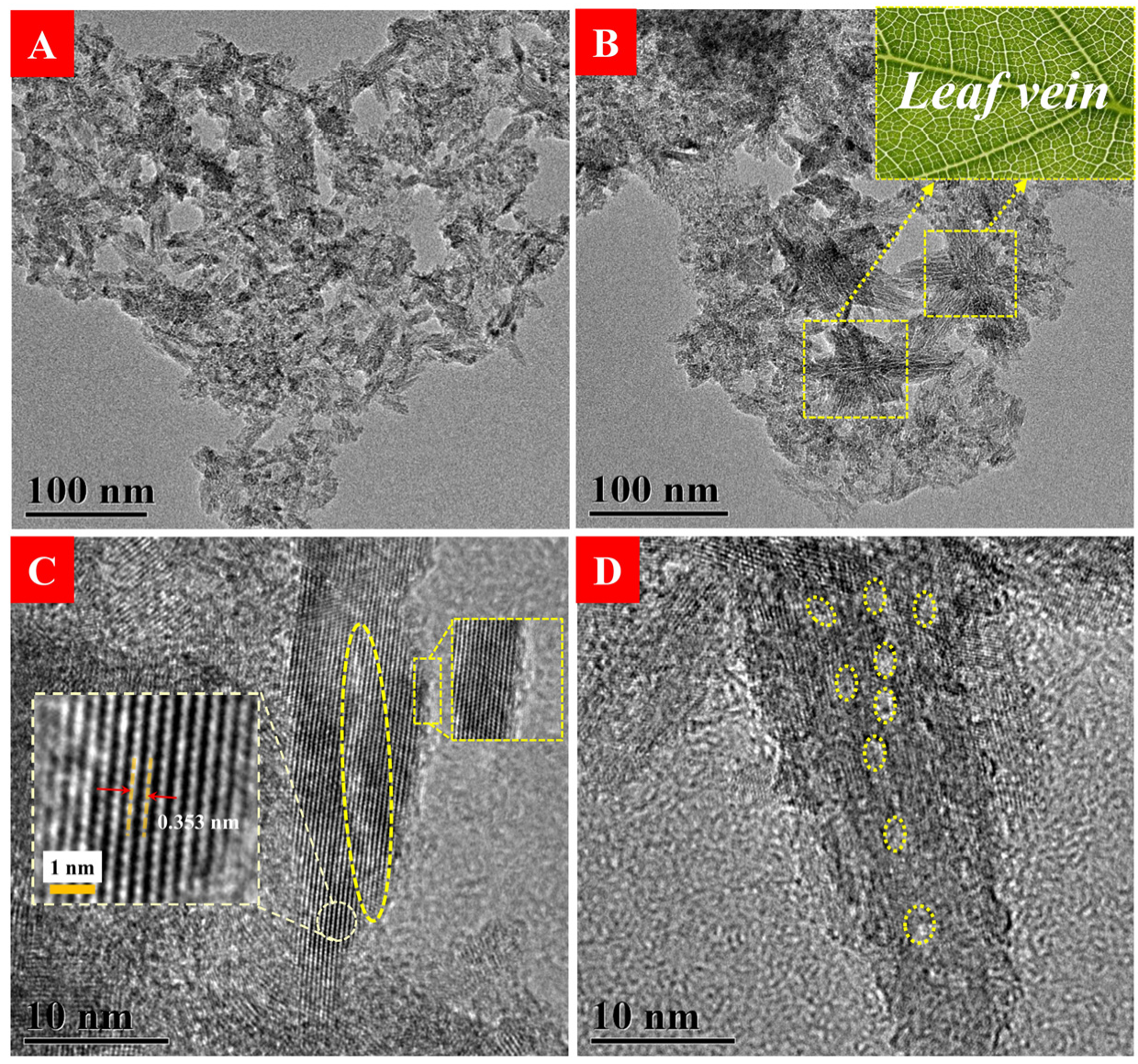
3.1. Characterization of HAm-TiO2−x
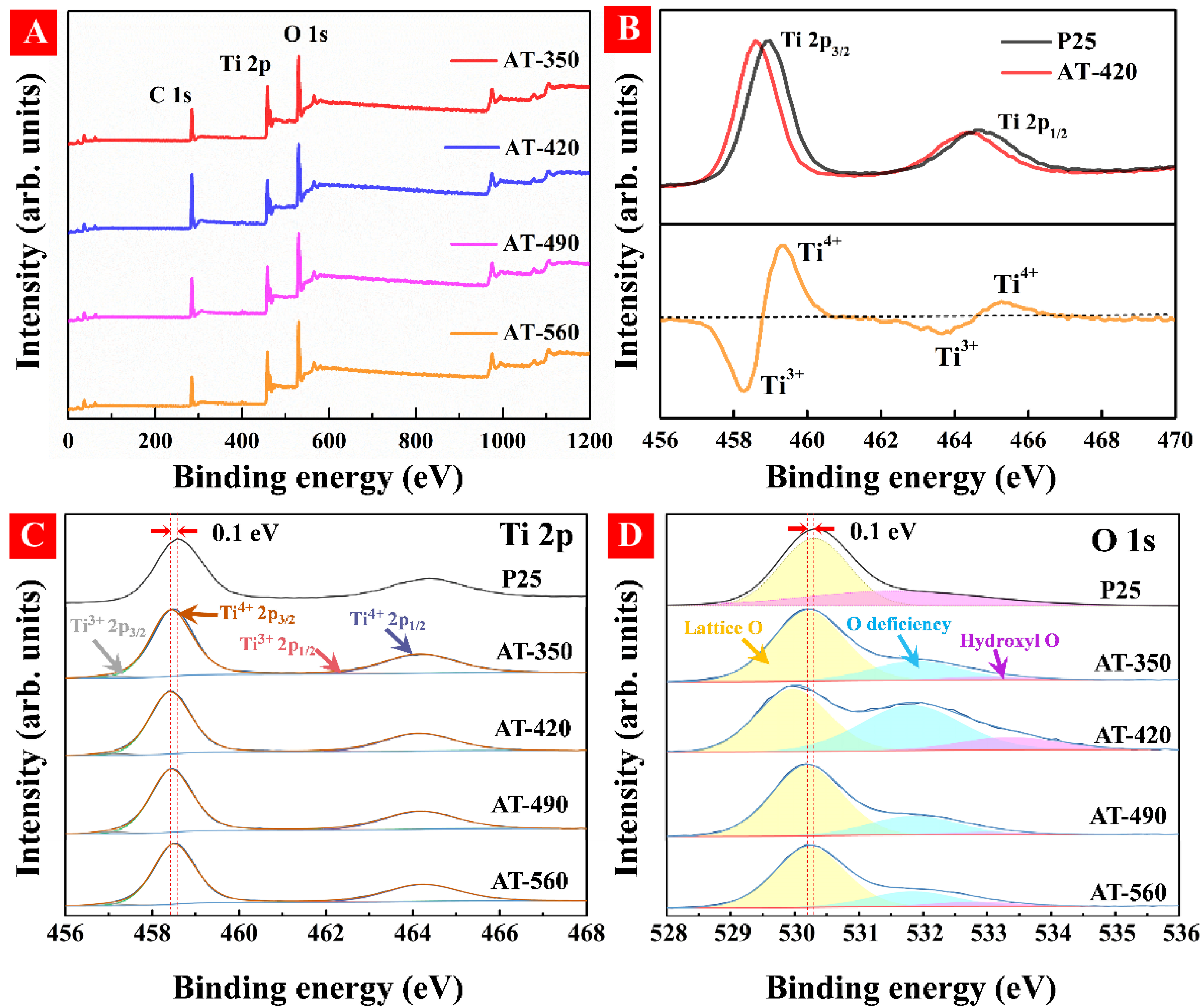
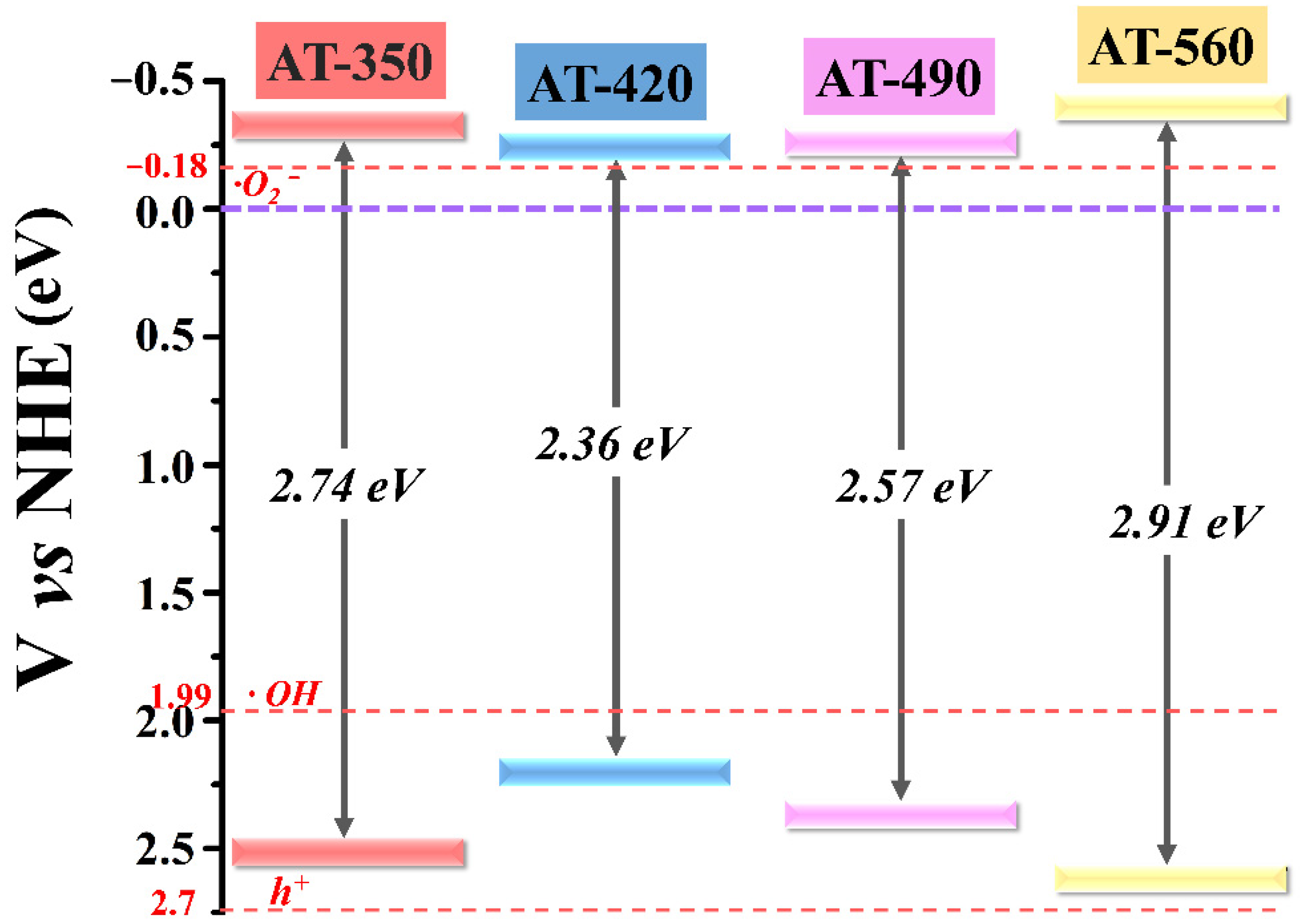
3.2. Regulation of Surface OV and Bandgap Engineering in HAm-TiO2−x
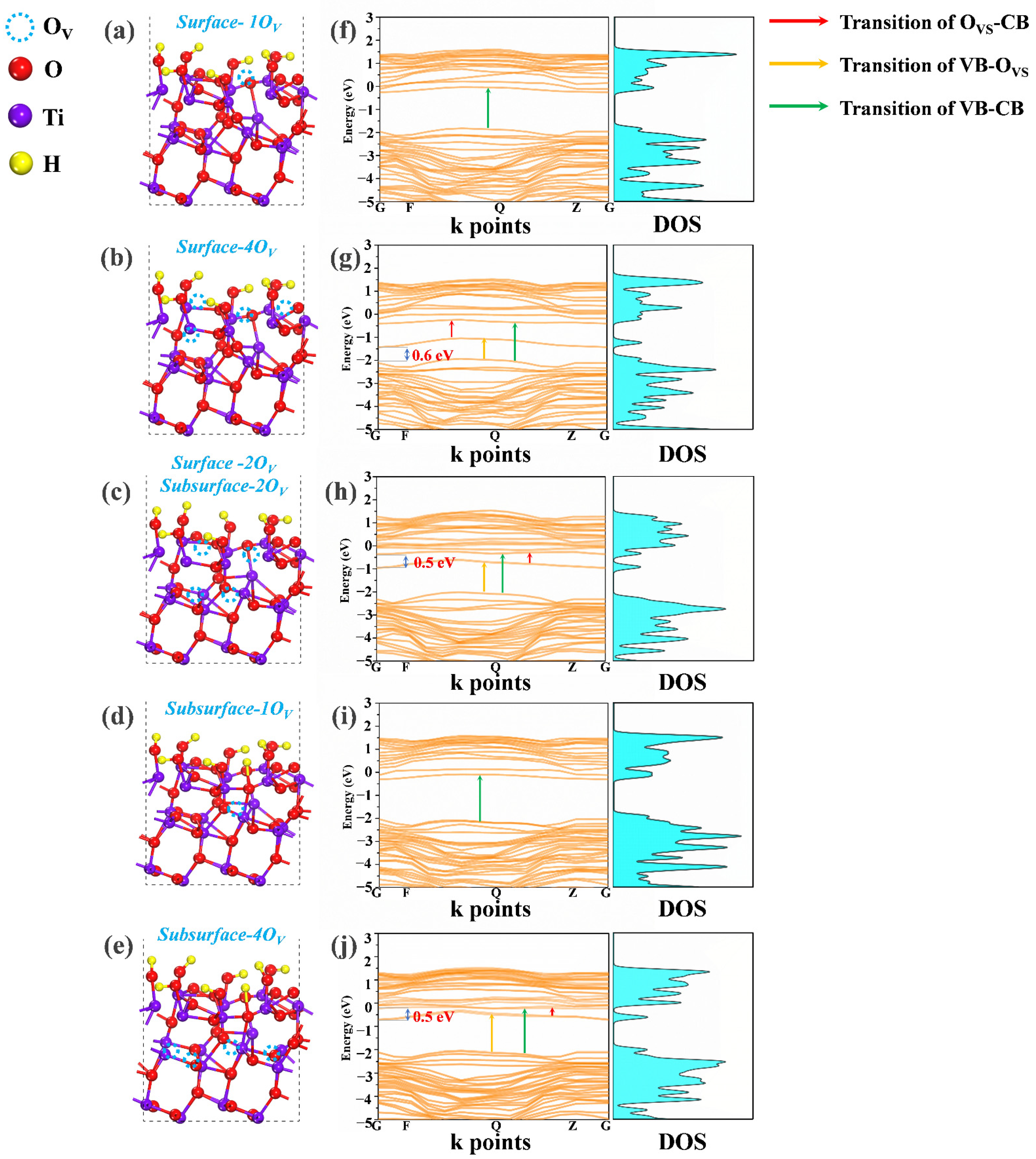
3.3. Effect of Distribution and Concentration of OV on Electrical Structure
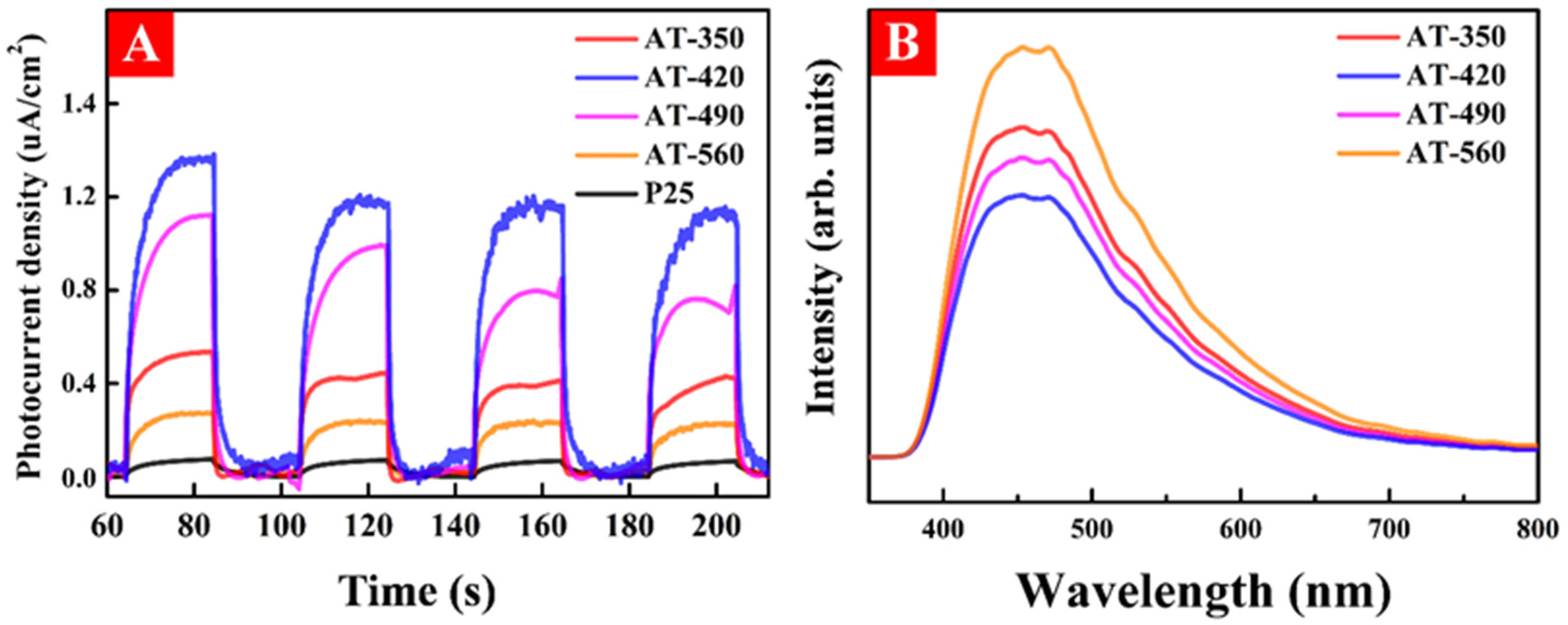
3.4. The Photo-Generated Carrier Activity of HAm-TiO2−x
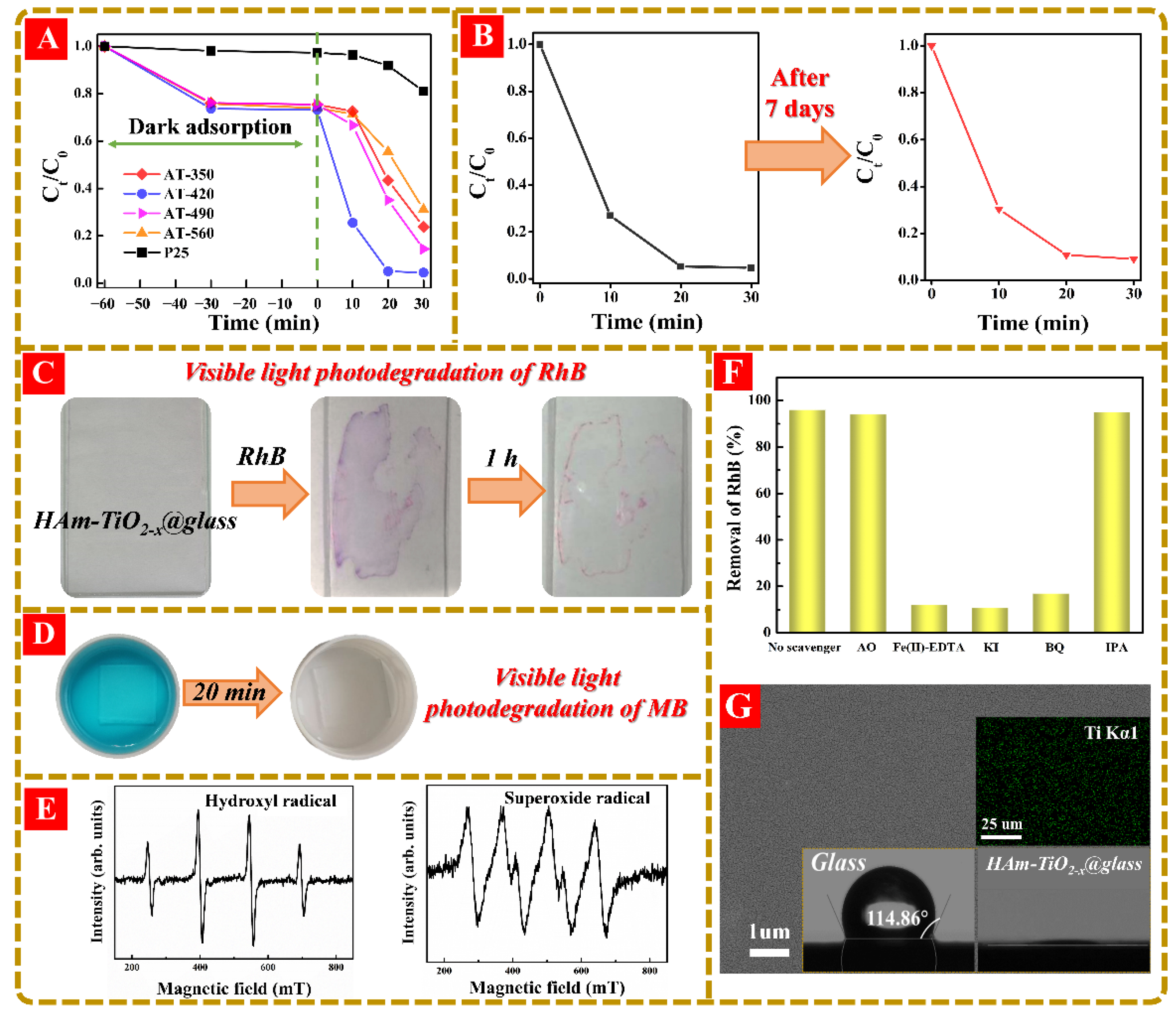
3.5. The Visible-Light-Driven Photodegradation of HAm-TiO2−x and Its Coatings
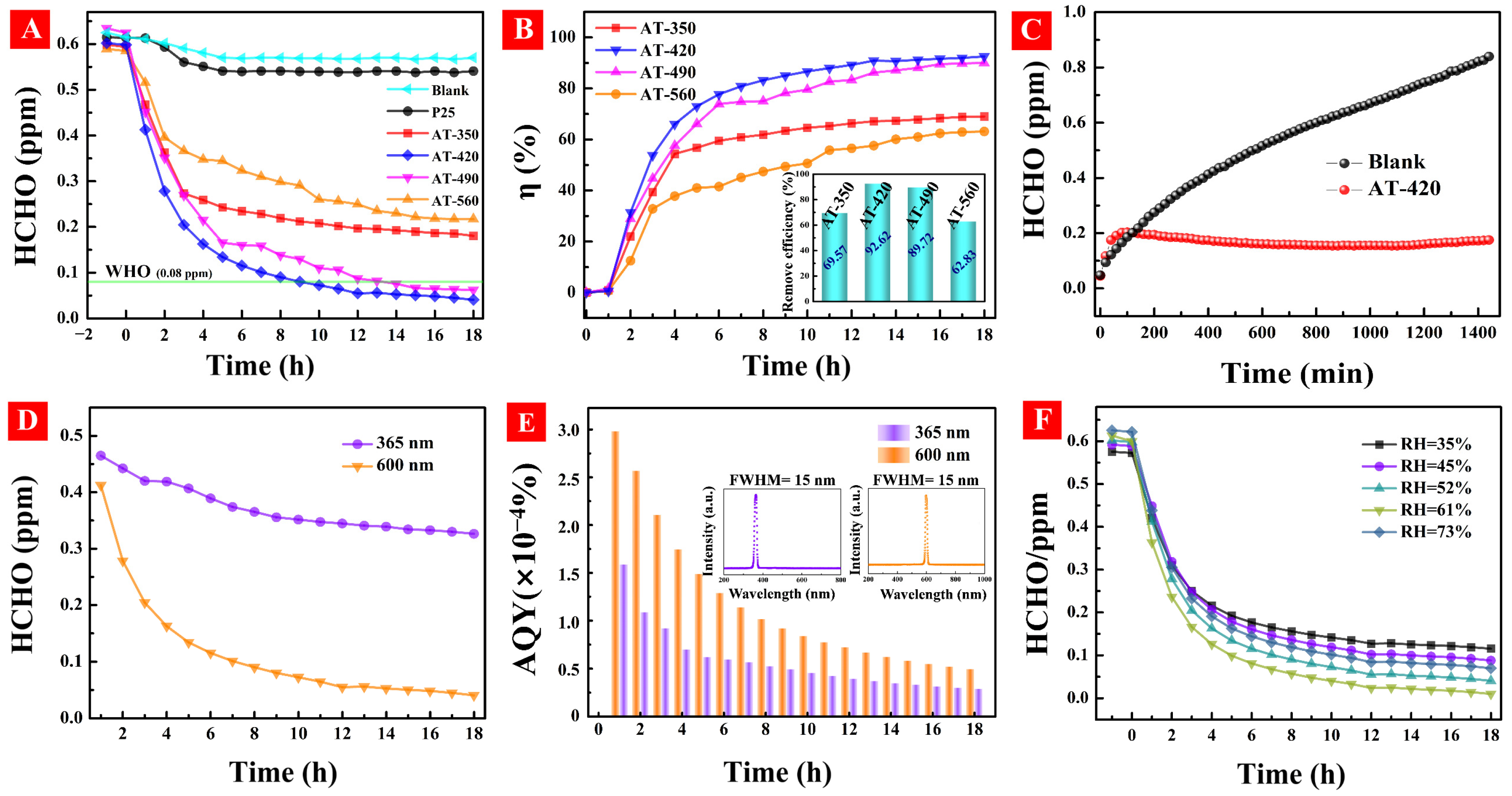
3.6. The HCHO Photodegradation Activity under Ambient Environment
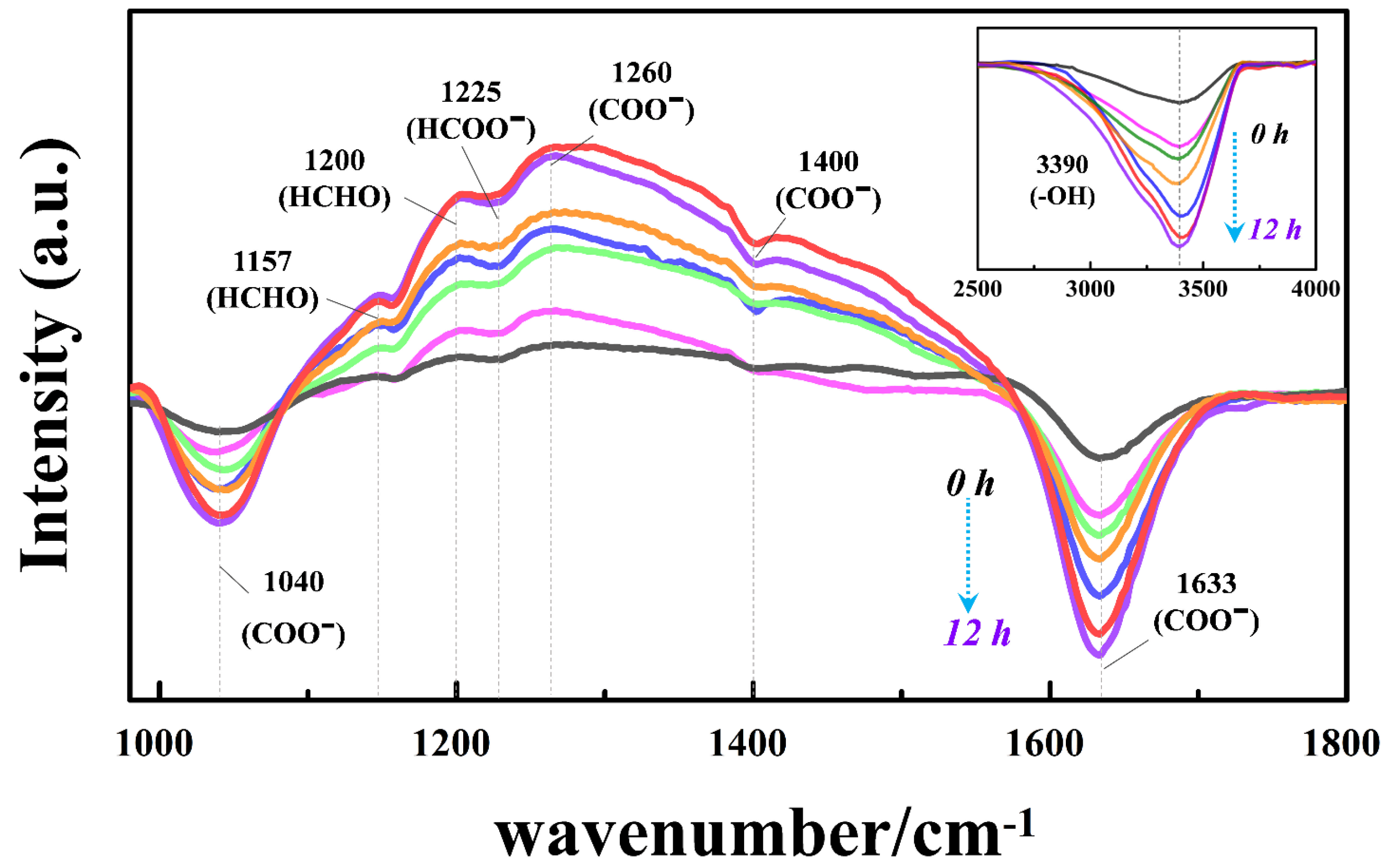
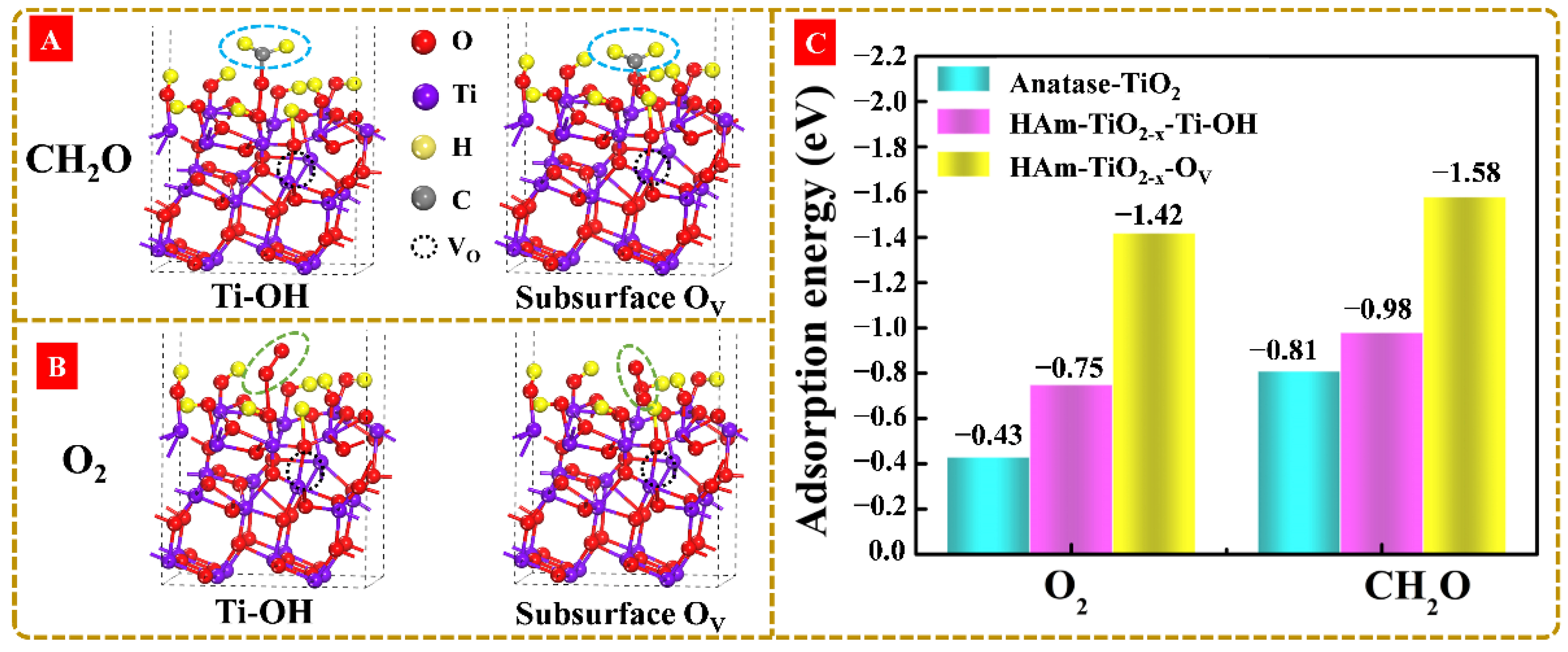
3.7. Investigations on the Surface Reaction Pathway and HCHO Adsorption Mechanism with the Subsurface OV
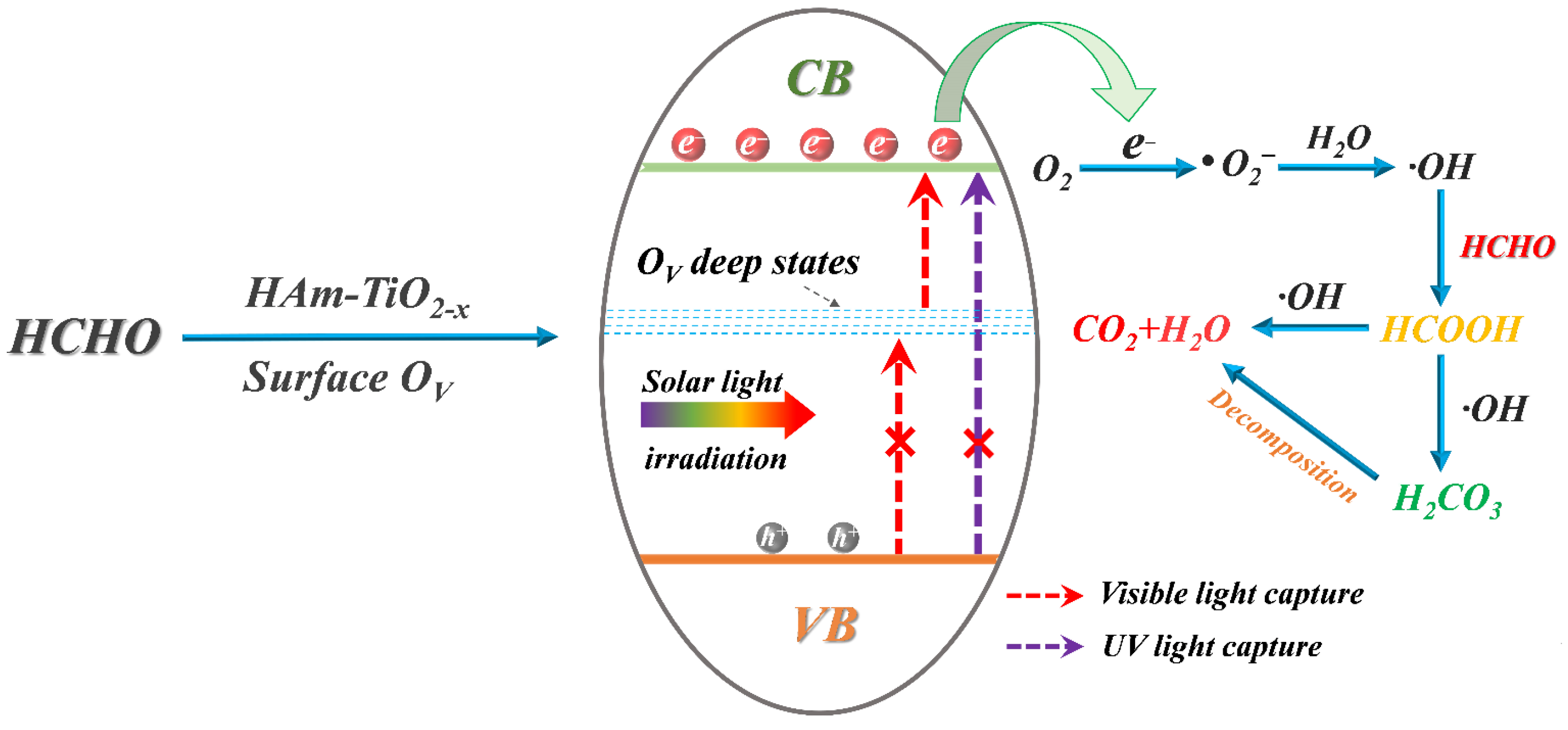
3.8. The Mechanism of HCHO Photocatalysis and OV Generation over HAm-TiO2−x
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fujishima, A.; Rao, T.N.; Tryk, D.A. Titanium dioxide photocatalysis. J. Photoch. Photobiol. C 2000, 1, 1–21. [Google Scholar] [CrossRef]
- Rao, Z.; Lu, G.; Mahmood, A.; Shi, G.; Xie, X.; Sun, J. Deactivation and activation mechanism of TiO2 and rGO/Er3+-TiO2 during flowing gaseous VOCs photodegradation. Appl. Catal. B Environ. 2021, 284, 119813. [Google Scholar] [CrossRef]
- Daghrir, R.; Drogui, P.; Robert, D. Modified TiO2 for environmental photocatalytic applications: A review. Ind. Eng. Chem. Res. 2013, 52, 3581–3599. [Google Scholar] [CrossRef]
- Luciani, G.; Imparato, C.; Vitiello, G. Photosensitive hybrid nanostructured materials: The big challenges for sunlight capture. Catalysts 2020, 10, 103. [Google Scholar] [CrossRef] [Green Version]
- Janczarek, M.; Endo-Kimura, M.; Raja-Mogan, T.; Kowalska, E. The role of oxygen vacancy and other defects for activity enhancement. In Green Photocatalytic Semiconductors: Recent Advances and Applications; Garg, S., Chandra, A., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 337–355. [Google Scholar]
- Hao, L.; Huang, H.; Zhang, Y.; Ma, T. Oxygen vacant semiconductor photocatalysts. Adv. Funct. Mater. 2021, 31, 2100919. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Z.; Li, G.; Huang, X.; Hao, W.; Bi, Y. Direct observation of oxygen vacancy self-healing on TiO2 photocatalysts for solar water splitting. Angew. Chem. Int. Edit. 2019, 58, 14229–14233. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, M.; Guan, Z.; Li, Q.; He, C.; Yang, J. Synergistic effect of surface and bulk single-electron-trapped oxygen vacancy of TiO2 in the photocatalytic reduction of CO2. Appl. Catal. B Environ. 2017, 206, 300–307. [Google Scholar] [CrossRef]
- Lin, L.; Ma, Y.; Wu, J.; Pang, F.; Ge, J.; Sui, S.; Yao, Y.; Qi, R.; Cheng, Y.; Duan, C.; et al. Origin of photocatalytic activity in Ti4+/Ti3+ core–shell titanium oxide nanocrystals. J. Phys. Chem. C 2019, 123, 20949–20959. [Google Scholar] [CrossRef]
- Mao, J.; An, X.; Gu, Z.; Zhou, J.; Liu, H.; Qu, J. Visualizing the interfacial charge transfer between photoactive microcystis aeruginosa and hydrogenated TiO2. Environ. Sci. Technol. 2020, 54, 10323–10332. [Google Scholar] [CrossRef]
- Li, J.; Weng, B.; Cai, S.; Chen, J.; Jia, H.; Xu, Y. Efficient promotion of charge transfer and separation in hydrogenated TiO2/WO3 with rich surface-oxygen-vacancies for photodecomposition of gaseous toluene. J. Hazard. Mater. 2018, 342, 661–669. [Google Scholar] [CrossRef]
- Yan, Y.; Cheng, X.; Zhang, W.; Chen, G.; Li, H.; Konkin, A.; Sun, Z.; Sun, S.; Wang, D.; Schaaf, P. Plasma hydrogenated TiO2/Nickel foam as an efficient bifunctional electrocatalyst for overall water splitting. ACS Sustain. Chem. Eng. 2019, 7, 885–894. [Google Scholar] [CrossRef]
- Zhang, K.; Park, J.H. Surface localization of defects in black TiO2: Enhancing photoactivity or reactivity. J. Phys. Chem. Lett. 2017, 8, 199–207. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Huang, F. Black titanium dioxide (TiO2) nanomaterials. Chem. Soc. Rev. 2015, 44, 1861–1885. [Google Scholar] [CrossRef]
- Wang, G.; Wang, H.; Ling, Y.; Tang, Y.; Yang, X.; Fitzmorris, R.C.; Wang, C.; Zhang, J.Z.; Li, Y. Hydrogen-treated TiO2 nanowire arrays for photoelectrochemical water splitting. Nano Lett. 2011, 11, 3026–3033. [Google Scholar] [CrossRef] [PubMed]
- Danyliuk, N.; Tatarchuk, T.; Kannan, K.; Shyichuk, A. Optimization of TiO2-P25 photocatalyst dose and H2O2 concentration for advanced photo-oxidation using smartphone-based colorimetry. Water Sci. Technol. 2021, 84, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Tatarchuk, T.; Danyliuk, N.; Shyichuk, A.; Macyk, W.; Naushad, M. Photocatalytic degradation of dyes using rutile TiO2 synthesized by reverse micelle and low temperature methods: Real-time monitoring of the degradation kinetics. J. Mol. Liq. 2021, 342, 117407. [Google Scholar] [CrossRef]
- Kang, J.; Zhang, Y.; Chai, Z.; Qiu, X.; Cao, X.; Zhang, P.; Teobaldi, G.; Liu, L.; Guo, L. Amorphous domains in black titanium dioxide. Adv. Mater. 2021, 33, 2100407. [Google Scholar] [CrossRef]
- Wang, B.; Biesold, G.M.; Zhang, M.; Lin, Z. Amorphous inorganic semiconductors for the development of solar cell, photoelectrocatalytic and photocatalytic applications. Chem. Soc. Rev. 2021, 50, 6914–6949. [Google Scholar] [CrossRef]
- Sun, S.; Song, P.; Cui, J.; Liang, S. Amorphous TiO2 nanostructures: Synthesis, fundamental properties and photocatalytic applications. Catal. Sci. Technol. 2019, 9, 4198–4215. [Google Scholar] [CrossRef]
- Cheng, L.; Li, B.; Cheng, Q.; Baldwin, A.N.; Shang, Y. Investigations of indoor air quality of large department store buildings in China based on field measurements. Build. Environ. 2017, 118, 128–143. [Google Scholar] [CrossRef]
- Chang, T.; Wang, J.; Lu, J.; Shen, Z.; Huang, Y.; Sun, J.; Xu, H.; Wang, X.; Ren, D.; Cao, J. Evaluation of indoor air pollution during decorating process and inhalation health risks in Xi’an, China: A case study. Aerosol Air Qual. Res. 2019, 19, 854–864. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Ji, J.; Liu, B.; Huang, H. Reduced TiO2 with tunable oxygen vacancies for catalytic oxidation of formaldehyde at room temperature. Appl. Surf. Sci. 2019, 473, 934–942. [Google Scholar] [CrossRef]
- Zhu, M.; Muhammad, Y.; Hu, P.; Wang, B.; Wu, Y.; Sun, X.; Tong, Z.; Zhao, Z. Enhanced interfacial contact of dopamine bridged melamine-graphene/TiO2 nano-capsules for efficient photocatalytic degradation of gaseous formaldehyde. Appl. Catal. B Environ. 2018, 232, 182–193. [Google Scholar] [CrossRef]
- Huang, Q.; Hu, Y.; Pei, Y.; Zhang, J.; Fu, M. In situ synthesis of TiO2@NH2-MIL-125 composites for use in combined adsorption and photocatalytic degradation of formaldehyde. Appl. Catal. B Environ. 2019, 259, 118106. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Wang, C.; Zhang, C.; He, H. Sodium Enhances Ir/TiO2 activity for catalytic oxidation of formaldehyde at ambient temperature. ACS Catal. 2018, 8, 11377–11385. [Google Scholar] [CrossRef]
- Li, X.; Li, H.; Huang, Y.; Cao, J.; Huang, T.; Li, R.; Zhang, Q.; Lee, S.-c.; Ho, W. Exploring the photocatalytic conversion mechanism of gaseous formaldehyde degradation on TiO2–x-OV surface. J. Hazard. Mater. 2022, 424, 127217. [Google Scholar] [CrossRef]
- He, F.; Jeon, W.; Choi, W. Photocatalytic air purification mimicking the self-cleaning process of the atmosphere. Nature Commun. 2021, 12, 2528. [Google Scholar] [CrossRef]
- Wang, X.; Hong, S.; Lian, H.; Zhan, X.; Cheng, M.; Huang, Z.; Manzo, M.; Cai, L.; Nadda, A.; Le, Q.V.; et al. Photocatalytic degradation of surface-coated tourmaline-titanium dioxide for self-cleaning of formaldehyde emitted from furniture. J. Hazard. Mater. 2021, 420, 126565. [Google Scholar] [CrossRef]
- Deng, X.-Q.; Zhu, X.; Sun, Z.-G.; Li, X.-S.; Liu, J.-L.; Shi, C.; Zhu, A.-M. Exceptional activity for photocatalytic mineralization of formaldehyde over amorphous titania nanofilms. Chem. Eng. J. 2016, 306, 1001–1009. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, B.; Choudhury, A. Oxygen vacancy and dopant concentration dependent magnetic properties of Mn doped TiO2 nanoparticle. Curr. Appl. Phys. 2013, 13, 1025–1031. [Google Scholar] [CrossRef]
- Huang, H.; Hou, X.; Xiao, J.; Zhao, L.; Huang, Q.; Chen, H.; Li, Y. Effect of annealing atmosphere on the performance of TiO2 nanorod arrays in photoelectrochemical water splitting. Catal. Today 2019, 330, 189–194. [Google Scholar] [CrossRef]
- Dutta, S.; Chattopadhyay, S.; Jana, D.; Banerjee, A.; Manik, S.; Pradhan, S.K.; Sutradhar, M.; Sarkar, A. Annealing effect on nano-ZnO powder studied from positron lifetime and optical absorption spectroscopy. J. Appl. Phys. 2006, 100, 114328. [Google Scholar] [CrossRef]
- He, Y.; Dulub, O.; Cheng, H.; Selloni, A.; Diebold, U. Evidence for the predominance of subsurface defects on reduced anatase TiO2 (101). Phys. Rev. Lett. 2009, 102, 106105. [Google Scholar] [CrossRef]
- Sun, W.; Li, Y.; Shi, W.; Zhao, X.; Fang, P. Formation of AgI/TiO2 nanocomposite leads to excellent thermochromic reversibility and photostability. J. Mater. Chem. 2011, 21, 9263–9270. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, P.; Fan, Y.; Fan, Y.; Shi, X.; Cui, G.; Tang, B. Grey rutile TiO2 with long-term photocatalytic activity synthesized via two-step calcination. Nanomaterials 2020, 10, 920. [Google Scholar] [CrossRef]
- Huang, J.; Yang, K.; Zhang, Z.; Yang, L.; Hirano, S. Layered perovskite LiEuTiO4 as a 0.8 V lithium intercalation electrode. Chem. Commun. 2017, 53, 7800–7803. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Moon, B.M.; Lee, K.M.; Kim, Y.H.; Park, H.G.; Lim, J.H.; Oh, B.Y.; Kim, B.Y.; Hwang, J.Y.; Ok, C.H.; et al. Homogeneous liquid crystal orientation on ion beam exposure TiO2 surfaces depending on an anisotropic dipole field. Liq. Cryst. 2010, 37, 279–284. [Google Scholar] [CrossRef]
- Shi, C.; Qi, H.; Sun, Z.; Qu, K.; Huang, Z.; Li, J.; Dong, M.; Guo, Z. Carbon dot-sensitized urchin-like Ti3+ self-doped TiO2 photocatalysts with enhanced photoredox ability for highly efficient removal of Cr6+ and RhB. J. Mater. Chem. C 2020, 8, 2238–2247. [Google Scholar] [CrossRef]
- Li, G.; Lian, Z.; Li, X.; Xu, Y.; Wang, W.; Zhang, D.; Tian, F.; Li, H. Ionothermal synthesis of black Ti3+-doped single-crystal TiO2 as an active photocatalyst for pollutant degradation and H2 generation. J. Mater. Chem. A 2015, 3, 3748–3756. [Google Scholar] [CrossRef]
- Guillemot, F.; Porté, M.C.; Labrugère, C.; Baquey, C. Ti4+ to Ti3+ conversion of TiO2 uppermost layer by low-temperature vacuum annealing: Interest for titanium biomedical applications. J. Colloid Interf. Sci. 2002, 255, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Yu, J.C.; Ming, T.; Wang, J. A wide-spectrum-responsive TiO2 photoanode for photoelectrochemical cells. Appl. Catal. B Environ. 2015, 168–169, 483–489. [Google Scholar] [CrossRef]
- Qian, A.; Hyeon, S.E.; Seo, J.Y.; Chung, C.-H. Capacitance changes associated with cation-transport in free-standing flexible Ti3C2Tx (T=O, F, OH) MXene film electrodes. Electrochim. Acta 2018, 266, 86–93. [Google Scholar] [CrossRef]
- Qiu, H.; Ma, X.; Sun, C.; Zhao, B.; Chen, F. Surface oxygen vacancies enriched Pt/TiO2 synthesized with a defect migration strategy for superior photocatalytic activity. Appl. Surf. Sci. 2020, 506, 145021. [Google Scholar] [CrossRef]
- Xu, K.; Yang, X.; Sun, D.; Yang, X.; Zhou, Y.; Li, W.; Yang, Q.; Yang, X.; Li, R.; Feng, J. Enhanced visible-light driven photocatalytic performances over LaFeO3/NiO modified porous g-C3N4 nanosheets. Nano 2019, 15, 2050010. [Google Scholar] [CrossRef]
- Gu, Y.H.; Fujimiya, Y.; Kunugita, N. Long-term exposure to gaseous formaldehyde promotes allergen-specific IgE-mediated immune responses in a murine model. Hum. Exp. Toxicol. 2008, 27, 37–43. [Google Scholar] [CrossRef]
- Alminshid, A.H.; Abbas, M.N.; Alalwan, H.A.; Sultan, A.J.; Kadhom, M.A. Aldol condensation reaction of acetone on MgO nanoparticles surface: An in-situ drift investigation. Mol. Catal. 2021, 501, 111333. [Google Scholar] [CrossRef]
- Li, J.; Cui, W.; Chen, P.; Dong, X.A.; Chu, Y.; Sheng, J.; Zhang, Y.; Wang, Z.; Dong, F. Unraveling the mechanism of binary channel reactions in photocatalytic formaldehyde decomposition for promoted mineralization. Appl. Catal. B Environ. 2020, 260, 118130. [Google Scholar] [CrossRef]
- Jodłowski, P.J.; Jędrzejczyk, R.J.; Chlebda, D.; Gierada, M.; Łojewska, J. In situ spectroscopic studies of methane catalytic combustion over Co, Ce, and Pd mixed oxides deposited on a steel surface. J. Catal. 2017, 350, 1–12. [Google Scholar] [CrossRef]
- Song, S.; Lu, C.; Wu, X.; Jiang, S.; Sun, C.; Le, Z. Strong base g-C3N4 with perfect structure for photocatalytically eliminating formaldehyde under visible-light irradiation. Appl. Catal. B Environ. 2018, 227, 145–152. [Google Scholar] [CrossRef]
- Lu, J.; Zhong, J.; Ren, Q.; Li, J.; Song, L.; Mo, S.; Zhang, M.; Chen, P.; Fu, M.; Ye, D. Construction of Cu-Ce interface for boosting toluene oxidation: Study of Cu-Ce interaction and intermediates identified by in situ DRIFTS. Chin. Chem. Lett. 2021, 32, 3435–3439. [Google Scholar] [CrossRef]
- Xu, P.; Xu, T.; Yu, H.; Zheng, D.; Li, X. MOF (metal-organic framework) nanomaterial for 400 ppb-concentration detectable xylene gas sensors. In Proceedings of the 2017 IEEE 30th International Conference on Micro Electro Mechanical Systems (MEMS), Las Vegas, NV, USA, 22–26 January 2017; pp. 1075–1078. [Google Scholar]
- Hu, Z.; Yang, C.; Lv, K.; Li, X.; Li, Q.; Fan, J. Single atomic Au induced dramatic promotion of the photocatalytic activity of TiO2 hollow microspheres. Chem. Commun. 2020, 56, 1745–1748. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zheng, J.; Qiu, S.; Wu, M.; Zhang, Q.; Yan, Z.; Xue, Q. Review on electrical discharge plasma technology for wastewater remediation. Chem. Eng. J. 2014, 236, 348–368. [Google Scholar] [CrossRef]
- Oinuma, G.; Nayak, G.; Du, Y.; Bruggeman, P.J. Controlled plasma—Droplet interactions: A quantitative study of OH transfer in plasma–liquid interaction. Plasma Sources Sci. Technol. 2020, 29, 095002. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | τ1 (ns) | OVL-I1-% | τ2 (ns) | OVS-I2-% | τ3 (ns) | LV-I3-% |
|---|---|---|---|---|---|---|
| AT-350 | 0.323 | 77.1 | 0.453 | 22.6 | 5.142 | 0.3 |
| AT-420 | 0.224 | 10.8 | 0.363 | 89.2 | / | / |
| AT-490 | 0.247 | 15.4 | 0.369 | 84.6 | / | / |
| AT-560 | 0.329 | 87.8 | 0.526 | 11.8 | 3.797 | 0.4 |
| Sample | AT-350 | AT-420 | AT-490 | AT-560 |
|---|---|---|---|---|
| Output power (W) | 350 | 420 | 490 | 560 |
| Bandgap (eV) | 2.74 | 2.36 | 2.57 | 2.91 |
| VBM (eV) | 2.48 | 2.16 | 2.33 | 2.58 |
| Ratio of Ti3+/Ti4+ | 2.81% | 3.01% | 2.91% | 2.69% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, G.; Hu, M.; Wu, B.; Shi, S.; Yuan, S.; Li, Y.; Zeng, H. Hydrogenated Amorphous Titania with Engineered Surface Oxygen Vacancy for Efficient Formaldehyde and Dye Removals under Visible-Light Irradiation. Nanomaterials 2022, 12, 742. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12050742
Feng G, Hu M, Wu B, Shi S, Yuan S, Li Y, Zeng H. Hydrogenated Amorphous Titania with Engineered Surface Oxygen Vacancy for Efficient Formaldehyde and Dye Removals under Visible-Light Irradiation. Nanomaterials. 2022; 12(5):742. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12050742
Chicago/Turabian StyleFeng, Guang, Mengyun Hu, Botao Wu, Shencheng Shi, Shuai Yuan, Yanan Li, and Heping Zeng. 2022. "Hydrogenated Amorphous Titania with Engineered Surface Oxygen Vacancy for Efficient Formaldehyde and Dye Removals under Visible-Light Irradiation" Nanomaterials 12, no. 5: 742. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12050742