
Development of a Bifunctional Ti-Based Gas Diffusion Electrode for ORR and OER by One- and Two-Step Pt-Ir Electrodeposition

 , and
, and
Abstract
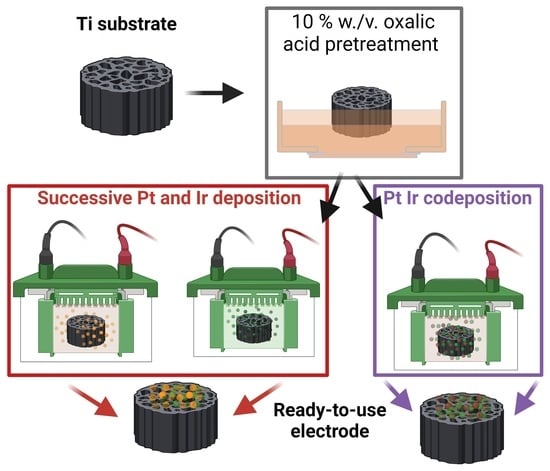
:
1. Introduction
2. Materials and Methods
2.1. Electrode Preparation
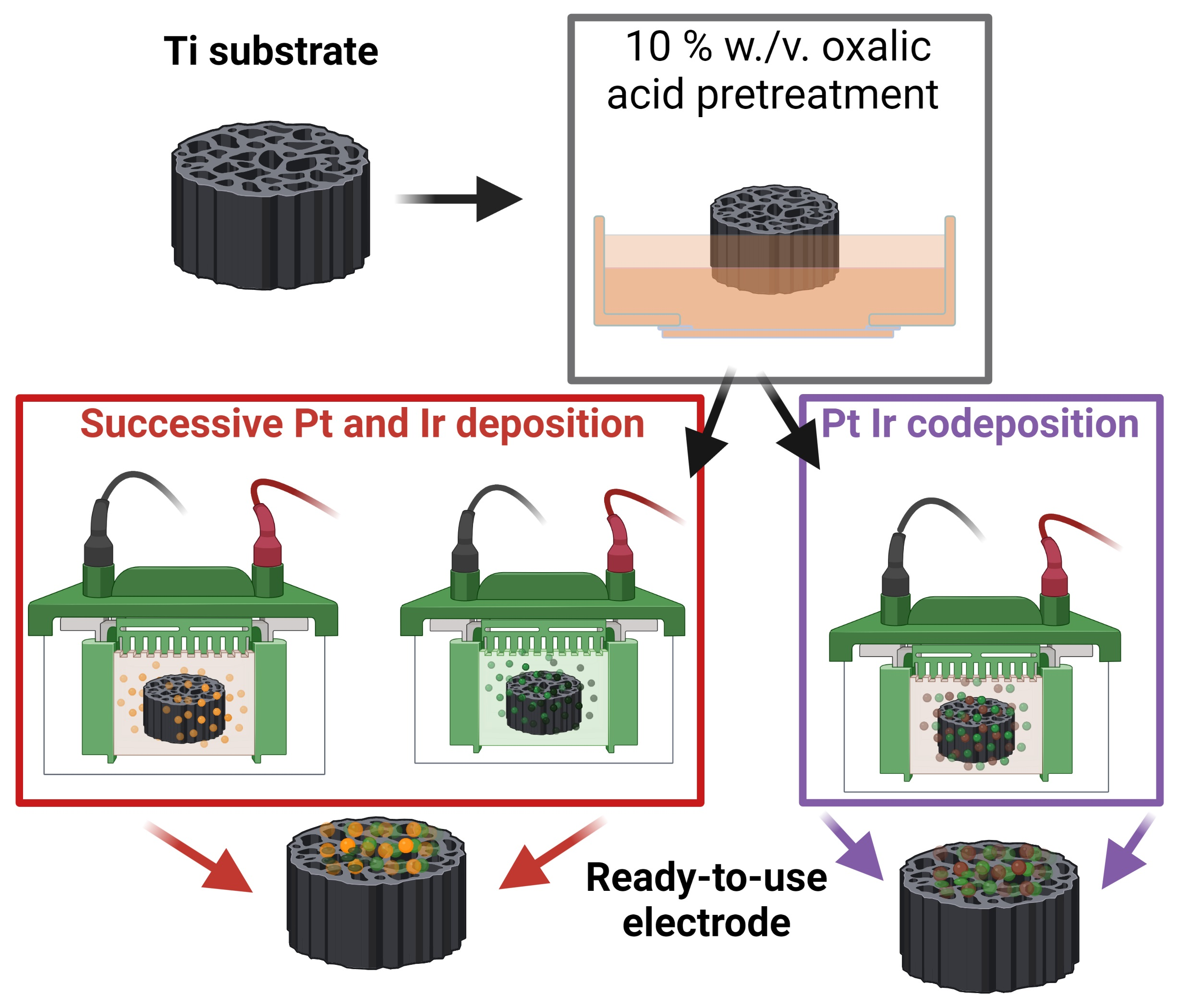
2.1.1. Substrate Cleaning
2.1.2. Electrochemical Catalyst Deposition
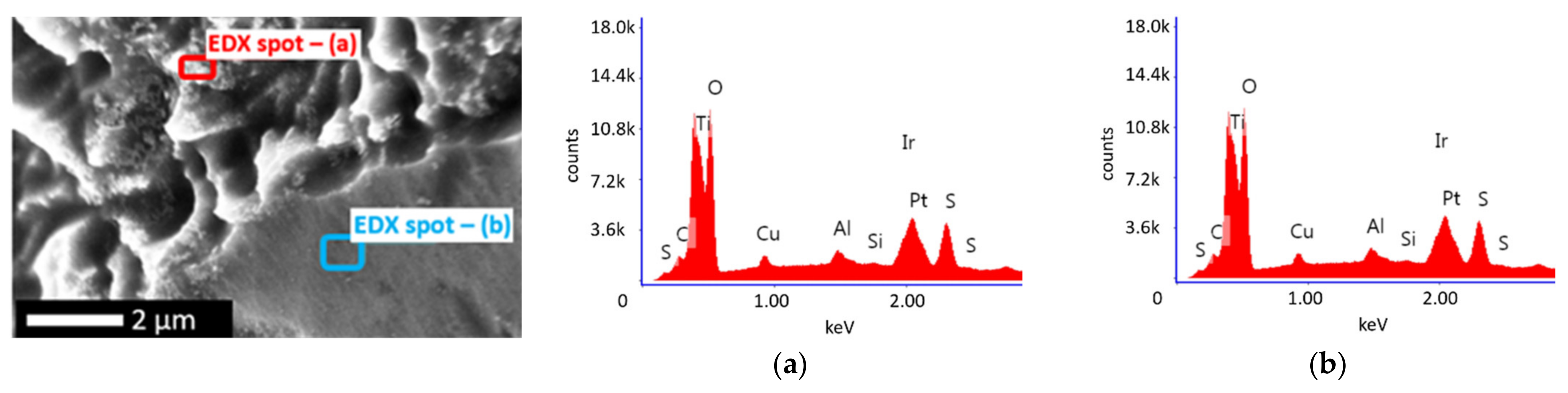
2.2. Physical Characterisation
2.3. Electrochemical Characterisation
3. Results and Discussion
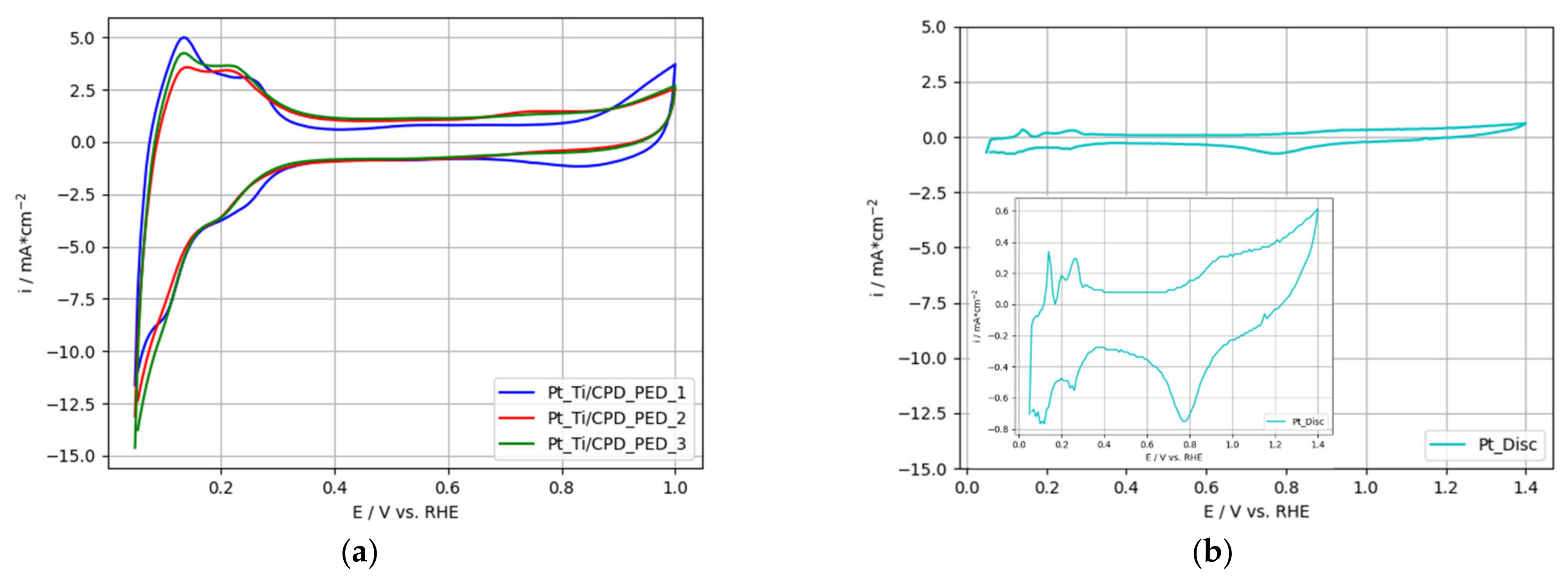
3.1. Ti-Based Electrodes with Single Pt Catalyst
3.2. Ti-Based Electrodes with Two-Step Deposited PtIr Catalyst
3.2.1. ORR-Activity Results for Two-Step Deposited PtIr Electrodes
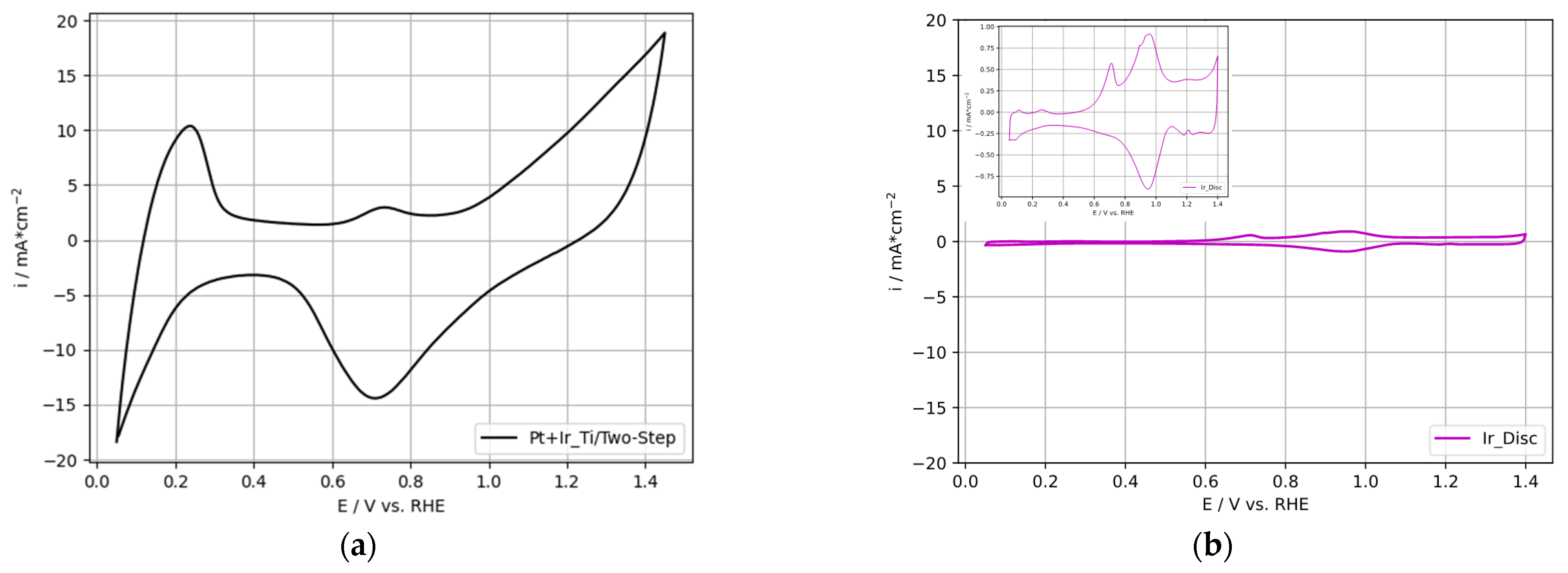
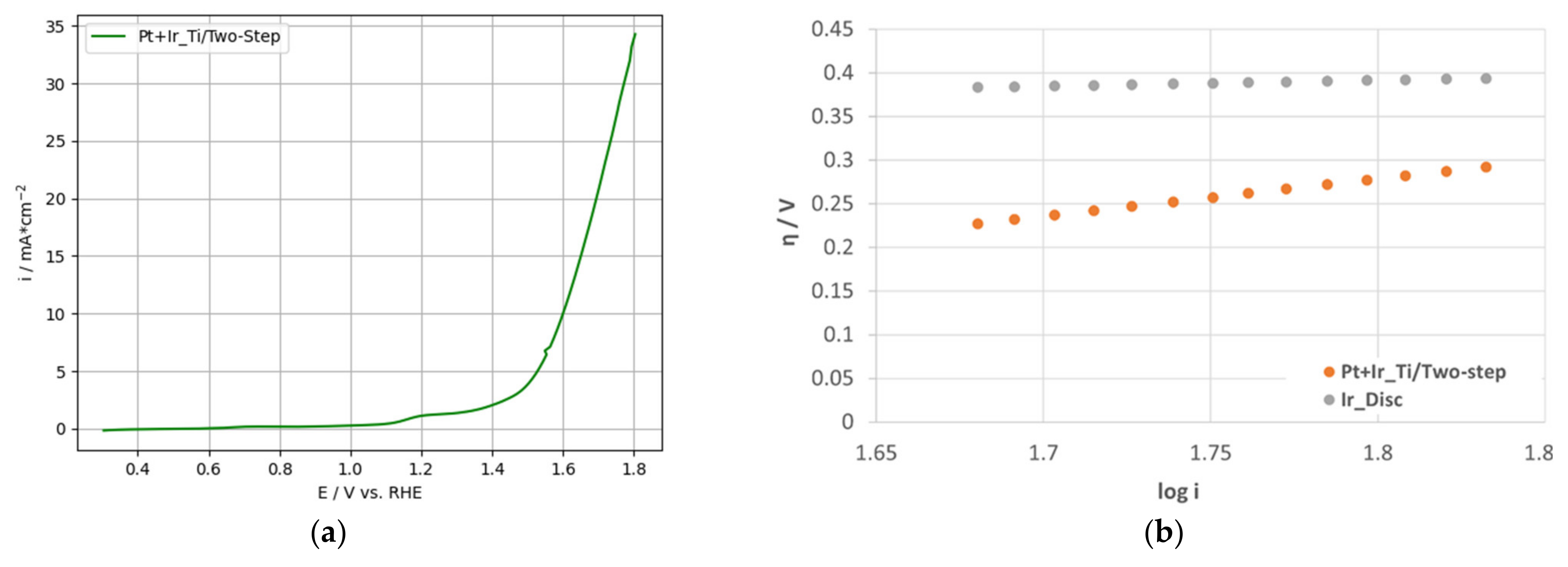
3.2.2. OER-Activity Results for Two-Step Deposited PtIr Electrodes
3.3. Ti-Based Electrodes with One-Step Deposited PtIr Catalyst
3.3.1. ORR-Activity for One-Step Deposited PtIr Electrodes
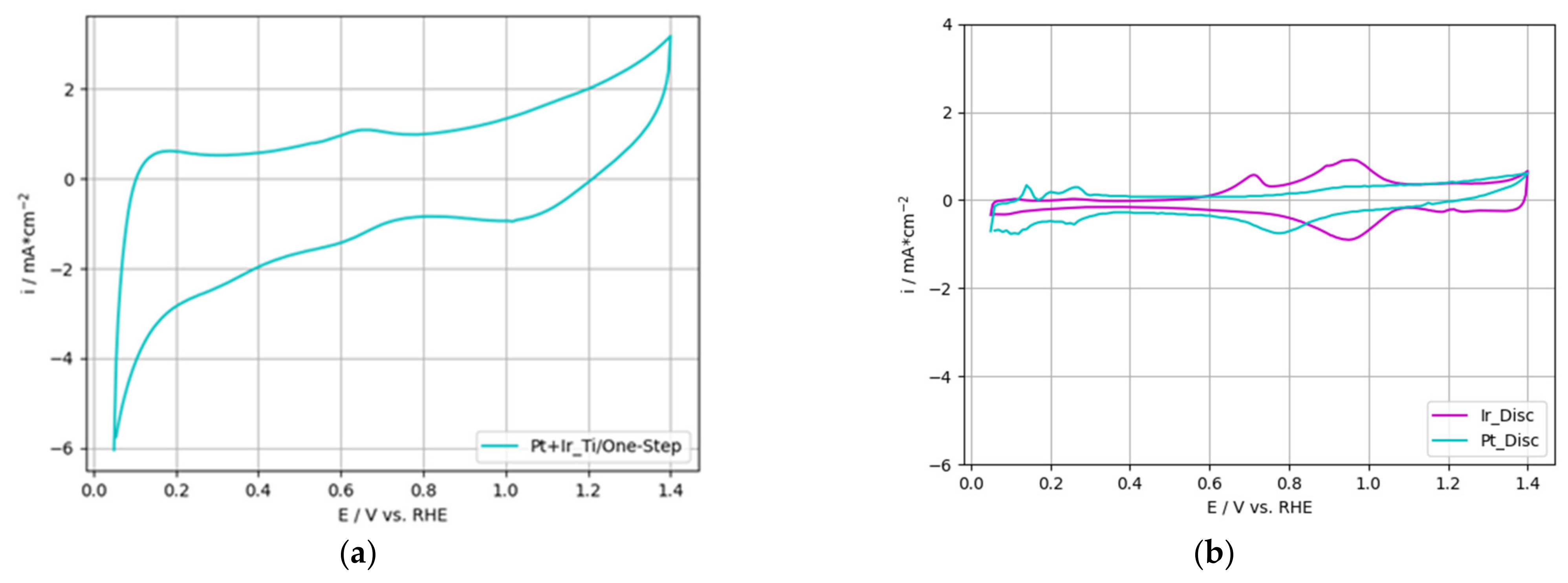
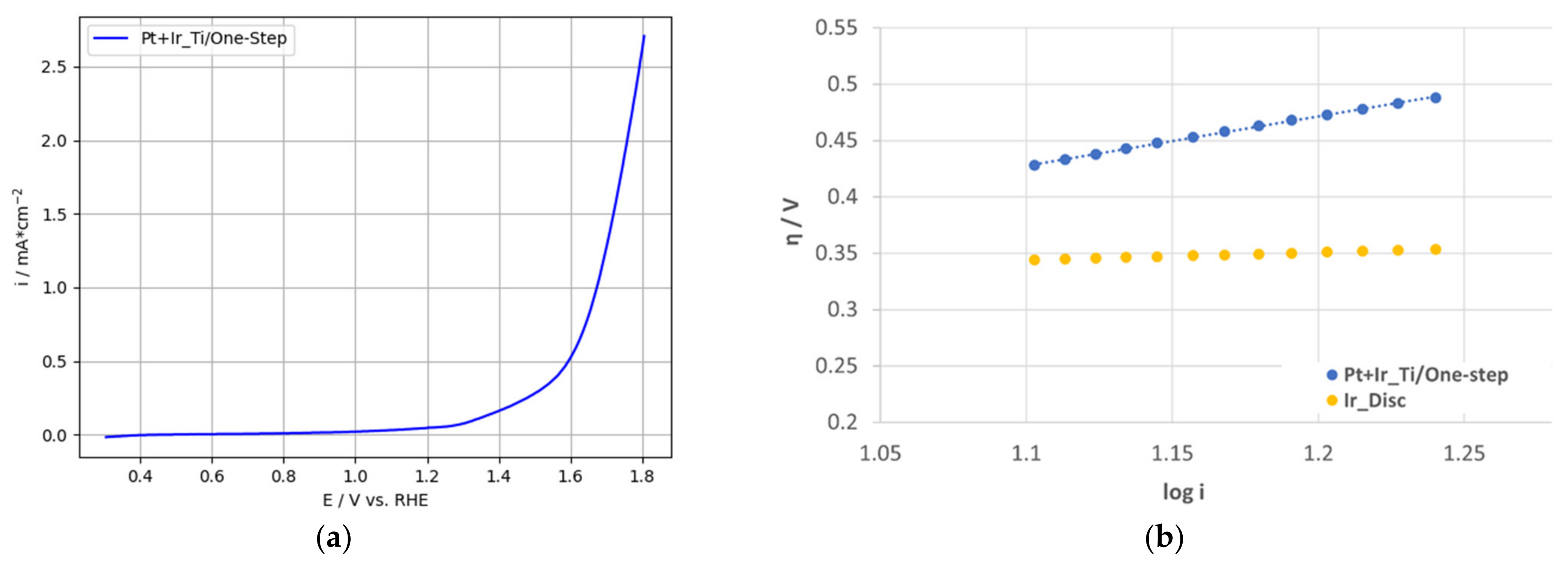
3.3.2. OER-Activity Results for One-Step Deposited PtIr Electrodes
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- United Nations. Paris Agreement; United Nations: Paris, France, 2015. [Google Scholar]
- Navigant, T.; Sach, K.; Jörling, B.; Lotz, M.; Jakob, H.; Schult, D. Klimaschutz in Zahlen: Fakten, Trends und Impulse Deutscher Klimapolitik; Bundesministerium für Umwelt, Naturschutz und nukleare Sicherheit: Berlin, Germany, 2020.
- Europäische Kommission. Förderung Einer Klimaneutralen Wirtschaft: Kommission Legt Pläne für das Energiesystem der Zukunft und Sauberen Wasserstoff Vor; European Commission: Brussels, Belgium, 2020. [Google Scholar]
- Dawood, F.; Anda, M.; Shafiullah, G.M. Hydrogen production for energy: An overview. Int. J. Hydrogen Energy 2020, 45, 3847–3869. [Google Scholar] [CrossRef]
- Sazali, N. Emerging technologies by hydrogen: A review. Int. J. Hydrogen Energy 2020, 45, 18753–18771. [Google Scholar] [CrossRef]
- Energiespeicher—Bedarf, Technologien, Integration; 2. Korrigierte und Ergänzte Auflage; Sterner, M.; Stadler, I. (Eds.) Springer Vieweg: Berlin, Germany, 2017; ISBN 978-3-662-48893-5. [Google Scholar]
- Hydrogen Council. Path to Hydrogen Competitivness: A Cost Perspective. 2020. Available online: https://www.h2knowledgecentre.com/content/policypaper1202?crawler=redirect&mimetype=application/pdf (accessed on 15 September 2021).
- Chapman, A.; Itaoka, K.; Hirose, K.; Davidson, F.T.; Nagasawa, K.; Lloyd, A.C.; Webber, M.E.; Kurban, Z.; Managi, S.; Tamaki, T.; et al. A review of four case studies assessing the potential for hydrogen penetration of the future energy system. Int. J. Hydrogen Energy 2019, 44, 6371–6382. [Google Scholar] [CrossRef]
- Paul, B.; Andrews, J. PEM unitised reversible/regenerative hydrogen fuel cell systems: State of the art and technical challenges. Renew. Sustain. Energy Rev. 2017, 79, 585–599. [Google Scholar] [CrossRef]
- Sadhasivam, T.; Dhanabalan, K.; Roh, S.-H.; Kim, T.-H.; Park, K.-W.; Jung, S.; Kurkuri, M.D.; Jung, H.-Y. A comprehensive review on unitized regenerative fuel cells: Crucial challenges and developments. Int. J. Hydrogen Energy 2017, 42, 4415–4433. [Google Scholar] [CrossRef]
- Hunsom, M.; Kaewsai, D.; Kannan, A.M. Recent developments in bifunctional air electrodes for unitized regenerative proton exchange membrane fuel cells—A review. Int. J. Hydrogen Energy 2018, 43, 21478–21501. [Google Scholar] [CrossRef]
- Mali, B.; Niraula, D.; Kafle, R.; Bhusal, A. Green hydrogen: Production methodology, applications and challenges in Nepal. In Proceedings of the 2021 7th International Conference on Engineering, Applied Sciences and Technology (ICEAST), Pattaya, Thailand, 1–3 April 2021; pp. 68–76, ISBN 978-1-6654-4122-3. [Google Scholar]
- Wang, Y.; Leung, D.Y.; Xuan, J.; Wang, H. A review on unitized regenerative fuel cell technologies, part-A: Unitized regenerative proton exchange membrane fuel cells. Renew. Sustain. Energy Rev. 2016, 65, 961–977. [Google Scholar] [CrossRef]
- Wang, Y.; Leung, D.Y.; Xuan, J.; Wang, H. A review on unitized regenerative fuel cell technologies, part B: Unitized regenerative alkaline fuel cell, solid oxide fuel cell, and microfluidic fuel cell. Renew. Sustain. Energy Rev. 2017, 75, 775–795. [Google Scholar] [CrossRef]
- Garche, J.; Smolinka, T.; Navarra, M.A.; Panero, S.; Scrosati, B. Regenerative fuel cells. In Electrochemical Power Sources: Fundamentals, Systems, and Applications; Elsevier: Amsterdam, The Netherlands, 2022; pp. 365–406. ISBN 9780128194249. [Google Scholar]
- Shiva Kumar, S.; Himabindu, V. Hydrogen production by PEM water electrolysis—A review. Mater. Sci. Energy Technol. 2019, 2, 442–454. [Google Scholar] [CrossRef]
- Töpler, J.; Lehmann, J. Wasserstoff und Brennstoffzelle; Springer: Berlin/Heidelberg, Germany, 2017; ISBN 978-3-662-53359-8. [Google Scholar]
- Lagadec, M.F.; Grimaud, A. Water electrolysers with closed and open electrochemical systems. Nat. Mater. 2020, 19, 1140–1150. [Google Scholar] [CrossRef]
- U.S. DOE Hydrogen and Fuel Cells Program Record. Fudel Cell System Cost—2015: Record #: 15015. Available online: https://www.hydrogen.energy.gov/pdfs/15015_fuel_cell_system_cost_2015.pdf (accessed on 20 October 2021).
- Schmidt, O.; Gambhir, A.; Staffell, I.; Hawkes, A.; Nelson, J.; Few, S. Future cost and performance of water electrolysis: An expert elicitation study. Int. J. Hydrogen Energy 2017, 42, 30470–30492. [Google Scholar] [CrossRef]
- Ayers, K.E.; Renner, J.N.; Danilovic, N.; Wang, J.X.; Zhang, Y.; Maric, R.; Yu, H. Pathways to ultra-low platinum group metal catalyst loading in proton exchange membrane electrolyzers. Catal. Today 2016, 262, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Hasran, U.A.; Pauzi, A.M.; Basri, S.; Karim, A.N. Recent perspectives and crucial challenges on unitized regenerative fuel cell (URFC). J. Kejuruter. 2018, 1, 37–46. [Google Scholar] [CrossRef]
- Yu, H.; Bonville, L.; Jankovic, J.; Maric, R. Microscopic insights on the degradation of a PEM water electrolyzer with ultra-low catalyst loading. Appl. Catal. B Environ. 2020, 260, 118194. [Google Scholar] [CrossRef]
- Jiang, G.; Yu, H.; Li, Y.; Yao, D.; Chi, J.; Sun, S.; Shao, Z. Low-loading and highly stable membrane electrode based on an Ir@WOxNR ordered array for PEM water electrolysis. ACS Appl. Mater. Interfaces 2021, 13, 15073–15082. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.; Kim, J.; Lee, H.J.; Kim, H.; Yoo, S.J.; Jang, J.H.; Young Park, H.; Sung, Y.-E.; Park, H.S. Low-loading IrO2 supported on Pt for catalysis of PEM water electrolysis and regenerative fuel cells. Appl. Catal. B Environ. 2020, 272, 118955. [Google Scholar] [CrossRef]
- Hegge, F.; Lombeck, F.; Cruz Ortiz, E.; Bohn, L.; von Holst, M.; Kroschel, M.; Hübner, J.; Breitwieser, M.; Strasser, P.; Vierrath, S. Efficient and stable low iridium loaded anodes for PEM water electrolysis made possible by nanofiber interlayers. ACS Appl. Energy Mater. 2020, 3, 8276–8284. [Google Scholar] [CrossRef]
- Lim, A.; Lee, J.S.; Lee, S.; Lee, S.Y.; Kim, H.; Yoo, S.J.; Jang, J.H.; Sung, Y.-E.; Park, H.S. Polymer electrolyte membrane unitized regenerative fuel cells: Operational considerations for achieving high round trip efficiency at low catalyst loading. Appl. Catal. B Environ. 2021, 297, 120458. [Google Scholar] [CrossRef]
- Wittstadt, U.; Wagner, E.; Jungmann, T. Membrane electrode assemblies for unitised regenerative polymer electrolyte fuel cells. J. Power Sources 2005, 145, 555–562. [Google Scholar] [CrossRef]
- Hwang, C.M.; Ishida, M.; Ito, H.; Maeda, T.; Nakano, A.; Kato, A.; Yoshida, T. Effect of titanium powder loading in gas diffusion layer of a polymer electrolyte unitized reversible fuel cell. J. Power Sources 2012, 202, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Hwang, C.M.; Ishida, M.; Ito, H.; Maeda, T.; Nakano, A.; Hasegawa, Y.; Yokoi, N.; Kato, A.; Yoshida, T. Influence of properties of gas diffusion layers on the performance of polymer electrolyte-based unitized reversible fuel cells. Int. J. Hydrogen Energy 2011, 36, 1740–1753. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-Y.; Ganesan, P.; Jung, H.-Y.; Popov, B.N. Development of supported bifunctional oxygen electrocatalysts and corrosion-resistant gas diffusion layer for unitized regenerative fuel cell applications. J. Power Sources 2012, 198, 23–29. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, H.; Zhong, H.; Ma, H. Gas diffusion layer with titanium carbide for a unitized regenerative fuel cell. Electrochim. Acta 2010, 55, 8801–8807. [Google Scholar] [CrossRef]
- Grigoriev, S.A.; Millet, P.; Dzhus, K.A.; Middleton, H.; Saetre, T.O.; Fateev, V.N. Design and characterization of bi-functional electrocatalytic layers for application in PEM unitized regenerative fuel cells. Int. J. Hydrogen Energy 2010, 35, 5070–5076. [Google Scholar] [CrossRef]
- Wu, X.; Tang, C.; Cheng, Y.; Min, X.; Jiang, S.P.; Wang, S. Bifunctional catalysts for reversible oxygen evolution reaction and oxygen reduction reaction. Chemistry 2020, 26, 3906–3929. [Google Scholar] [CrossRef] [PubMed]
- Retuerto, M.; Calle-Vallejo, F.; Pascual, L.; Lumbeeck, G.; Fernandez-Diaz, M.T.; Croft, M.; Gopalakrishnan, J.; Peña, M.A.; Hadermann, J.; Greenblatt, M.; et al. La1.5Sr0.5NiMn0.5Ru0.5O6 double perovskite with enhanced ORR/OER bifunctional catalytic activity. ACS Appl. Mater. Interfaces 2019, 11, 21454–21464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chala, S.A.; Tsai, M.-C.; Su, W.-N.; Ibrahim, K.B.; Duma, A.D.; Yeh, M.-H.; Wen, C.-Y.; Yu, C.-H.; Chan, T.-S.; Dai, H.; et al. Site activity and population engineering of NiRu-layered double hydroxide nanosheets decorated with silver nanoparticles for oxygen evolution and reduction reactions. ACS Catal. 2019, 9, 117–129. [Google Scholar] [CrossRef]
- Zhang, G.; Shao, Z.-G.; Lu, W.; Li, G.; Liu, F.; Yi, B. One-pot synthesis of Ir@Pt nanodendrites as highly active bifunctional electrocatalysts for oxygen reduction and oxygen evolution in acidic medium. Electrochem. Commun. 2012, 22, 145–148. [Google Scholar] [CrossRef]
- Siegmund, D.; Metz, S.; Peinecke, V.; Warner, T.E.; Cremers, C.; Grevé, A.; Smolinka, T.; Segets, D.; Apfel, U.-P. Crossing the Valley of Death: From Fundamental to Applied Research in Electrolysis. JACS Au 2021, 1, 527–535. [Google Scholar] [CrossRef]
- Du, J.; Quinson, J.; Zhang, D.; Bizzotto, F.; Zana, A.; Arenz, M. Bifunctional Pt-IrO2 Catalysts for the Oxygen Evolution and Oxygen Reduction Reactions: Alloy Nanoparticles vs. Nanocomposite Catalysts. ACS Catal. 2021, 11, 820–828. [Google Scholar] [CrossRef]
- Jung, H.-Y.; Park, S.; Popov, B.N. Electrochemical studies of an unsupported PtIr electrocatalyst as a bifunctional oxygen electrode in a unitized regenerative fuel cell. J. Power Sources 2009, 191, 357–361. [Google Scholar] [CrossRef]
- Cruz, J.C.; Rivas, S.; Beltran, D.; Meas, Y.; Ornelas, R.; Osorio-Monreal, G.; Ortiz-Frade, L.; Ledesma-García, J.; Arriaga, L.G. Synthesis and evaluation of ATO as a support for Pt–IrO2 in a unitized regenerative fuel cell. Int. J. Hydrogen Energy 2012, 37, 13522–13528. [Google Scholar] [CrossRef]
- Rost, U. Effect of process parameters for oxygen plasma activation of carbon nanofibers on the characteristics of deposited platinum nanoparticles as electrocatalyst in proton exchange membrane fuel cells. Int. J. Electrochem. Sci. 2016, 11, 9110–9122. [Google Scholar] [CrossRef]
- Rost, U.; Podleschny, P.; Schumacher, M.; Muntean, R.; Pascal, D.T.; Mutascu, C.; Koziolek, J.; Marginean, G.; Brodmann, M. Long-term stable electrodes based on platinum electrocatalysts supported on titanium sintered felt for the use in PEM fuel cells. IOP Conf. Ser. Mater. Sci. Eng. 2018, 416, 12013. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W. Effect of argon plasma treatment on surface-treated titanium. Adv. Dent. Oral Health 2019, 11, 158–164. [Google Scholar] [CrossRef]
- Jun, J.; Shin, J.-H.; Dhayal, M. Surface state of TiO2 treated with low ion energy plasma. Appl. Surf. Sci. 2006, 252, 3871–3877. [Google Scholar] [CrossRef]
- Norasetthekul, S.; Park, P.Y.; Baik, K.H.; Lee, K.P.; Shin, J.H.; Jeong, B.S.; Shishodia, V.; Lambers, E.S.; Norton, D.P.; Pearton, S.J. Dry etch chemistries for TiO2 thin films. Appl. Surf. Sci. 2001, 185, 27–33. [Google Scholar] [CrossRef]
- Mahé, E.; Devilliers, D. Surface modification of titanium substrates for the preparation of noble metal coated anodes. Electrochim. Acta 2001, 46, 629–636. [Google Scholar] [CrossRef]
- Yeniyol, S.; Bölükbaşi, N.; Çakir, A.F.; Bilir, A.; Özdemir, T. Effects of surface modifications with oxalic acid etching and sandblasting on surface topography and biocompatibility of cpTi surfaces. Biotechnol. Biotechnol. Equip. 2013, 27, 3995–4001. [Google Scholar] [CrossRef] [Green Version]
- Arenas, L.F.; Kaishubayeva, N.; Ponce de León, C.; Walsh, F.C. Electrodeposition of platinum on 3D-printed titanium mesh to produce tailored, high area anodes. Trans. IMF 2020, 98, 48–52. [Google Scholar] [CrossRef]
- Mitzel, J. Gasdiffusionselektroden für Polymerelektrolytmembran-Brennstoffzellen Mittels Elektrochemischer Abscheidungsverfahren. Ph.D. Thesis, Universität des Saarlandes, Saarbrücken, Germany, 2012. [Google Scholar]
- Abraham, B.G.; Chetty, R. Influence of electrodeposition techniques and parameters towards the deposition of Pt electrocatalysts for methanol oxidation. J. Appl. Electrochem. 2021, 51, 503–520. [Google Scholar] [CrossRef]
- Garsany, Y.; Baturina, O.A.; Swider-Lyons, K.E.; Kocha, S.S. Experimental methods for quantifying the activity of platinum electrocatalysts for the oxygen reduction reaction. Anal. Chem. 2010, 82, 6321–6328. [Google Scholar] [CrossRef] [PubMed]
- Ioroi, T.; Yasuda, K. Platinum-iridium alloys as oxygen reduction electrocatalysts for polymer electrolyte fuel cells. J. Electrochem. Soc. 2005, 152, A1917. [Google Scholar] [CrossRef]
- Yarar Kaplan, B.; Haghmoradi, N.; Jamil, E.; Merino, C.; Alkan Gürsel, S. Platinum nanoparticles decorated carbon nanofiber hybrids as highly active electrocatalysts for polymer electrolyte membrane fuel cells. Int. J. Energy Res. 2020, 44, 10251–10261. [Google Scholar] [CrossRef]
- Böhm, L.; Näther, J.; Underberg, M.; Kazamer, N.; Holtkotte, L.; Rost, U.; Marginean, G.; Wirkert, F.; Brodmann, M.; Hülser, T.; et al. Pulsed electrodeposition of iridium catalyst nanoparticles on titanium suboxide supports for application in PEM electrolysis. Mater. Today Proc. 2021, 45, 4254–4259. [Google Scholar] [CrossRef]
- Petrossians, A.; Whalen, J.J., III; Weiland, J.D.; Mansfeld, F. Electrodeposition and characterization of thin-film platinum-iridium alloys for biological interfaces. J. Electrochem. Soc. 2011, 158, D269. [Google Scholar] [CrossRef]
- Geiger, S.; Kasian, O.; Shrestha, B.; Mingers, A.; Mayrhofer, K.J. Activity and stability of electrochemically and thermally treated iridium for the oxygen evolution reaction. J. Electrchemical Soc. 2016, 2016, F3123–F3138. [Google Scholar] [CrossRef] [Green Version]
- Cherevko, S.; Geiger, S.; Kasian, O.; Mingers, A.; Mayrhofer, K.J. Oxygen evolution activity and stability of iridium in acidic media. Part 1—Metallic iridium. J. Electroanal. Chem. 2016, 773, 69–78. [Google Scholar] [CrossRef]
- Topalov, G.; Ganske, G.; Lefterova, E.; Schnakenberg, U.; Slavcheva, E. Preparation and properties of thin Pt–Ir films deposited by dc magnetron co-sputtering. Int. J. Hydrogen Energy 2011, 36, 15437–15445. [Google Scholar] [CrossRef]
- Radev, I.; Topalov, G.; Lefterova, E.; Ganske, G.; Schnakenberg, U.; Tsotridis, G.; Slavcheva, E. Optimization of platinum/iridium ratio in thin sputtered films for PEMFC cathodes. Int. J. Hydrogen Energy 2012, 37, 7730–7735. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Yang, P.; Wang, D.; Xu, H.; Wang, B.; Meng, F.; Zhang, J.; An, M. Electrodeposition: Synthesis of advanced transition metal-based catalyst for hydrogen production via electrolysis of water. J. Energy Chem. 2021, 57, 547–566. [Google Scholar] [CrossRef]
- Kong, F.-D.; Zhang, S.; Yin, G.-P.; Zhang, N.; Wang, Z.-B.; Du, C.-Y. Pt/porous-IrO2 nanocomposite as promising electrocatalyst for unitized regenerative fuel cell. Electrochem. Commun. 2012, 14, 63–66. [Google Scholar] [CrossRef]
- Rakousky, C.G. Langzeitstabilität der Polymerelektrolyt-Wasserelektrolyse bei Reduziertem Iridiumgehalt; Forschungszentrum Jülich: Jülich, Germany, 2016; ISBN 978-3-95806-147-7. [Google Scholar]
- Alves, V.A.; Da Silva, L.A.; Boodts, J.; Trasatti, S. Kinetics and mechanism of oxygen evolution on IrO2-based electrodes containing Ti and Ce acidic solutions. Electrochim. Acta 1994, 39, 1585–1589. [Google Scholar] [CrossRef]
- Marshall, A.; Børresen, B.; Hagen, G.; Tsypkin, M.; Tunold, R. Electrochemical characterisation of IrxSn1−xO2 powders as oxygen evolution electrocatalysts. Electrochim. Acta 2006, 51, 3161–3167. [Google Scholar] [CrossRef]
- Antolini, E. Iridium as catalyst and cocatalyst for oxygen evolution/reduction in acidic polymer electrolyte membrane electrolyzers and fuel cells. ACS Catal. 2014, 4, 1426–1440. [Google Scholar] [CrossRef]
- Tahir, M.; Pan, L.; Idrees, F.; Zhang, X.; Wang, L.; Zou, J.-J.; Wang, Z.L. Electrocatalytic oxygen evolution reaction for energy conversion and storage: A comprehensive review. Nano Energy 2017, 37, 136–157. [Google Scholar] [CrossRef]
- Yan, Y.; Xia, B.Y.; Zhao, B.; Wang, X. A review on noble-metal-free bifunctional heterogeneous catalysts for overall electrochemical water splitting. J. Mater. Chem. A 2016, 4, 17587–17603. [Google Scholar] [CrossRef] [Green Version]
- Hatami, E.; Toghraei, A.; Barati Darband, G. Electrodeposition of Ni–Fe micro/nano urchin-like structure as an efficient electrocatalyst for overall water splitting. Int. J. Hydrogen Energy 2021, 46, 9394–9405. [Google Scholar] [CrossRef]
- Sheela, G.; Pushpavanam, M.; Pushpavanam, S. A bromide electrolyte for the electrodeposition of platinum and platinum–iridium alloy. Trans. IMF 2005, 83, 77–81. [Google Scholar] [CrossRef]
- García, G.; Roca-Ayats, M.; Lillo, A.; Galante, J.L.; Peña, M.A.; Martínez-Huerta, M.V. Catalyst support effects at the oxygen electrode of unitized regenerative fuel cells. Catal. Today 2013, 210, 67–74. [Google Scholar] [CrossRef]
- Da Silva, G.C.; Fernandes, M.R.; Ticianelli, E.A. Activity and stability of Pt/IrO2 bifunctional materials as catalysts for the oxygen evolution/reduction reactions. ACS Catal. 2018, 8, 2081–2092. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPD | PED | ||||||
|---|---|---|---|---|---|---|---|
| Noble Metal | U/V vs. CE | t/s | i/mA cm−2 | ton/s | toff/s | Cycles | |
| Two-step deposition | Pt | 1.8 | 450 | 40 | 0.01 | 0.056 | 4500 |
| Ir | / | / | 30 | 0.025 | 0.15 | 3000 | |
| One-step deposition | PtIr | / | / | 0.4 | 0.01 | 0.056 | 18,000 |
| QDesorp/ µC cmgeo.−2 | SRF/ cmechem.2 cmgeo.−2 | mPt/ µg cmgeo.−2 | ECSA/ mechem.2 g−1 | |
|---|---|---|---|---|
| Pt_Ti/CPD_PED_1 | 6104.4 | 29.1 | 262.5 | 11.1 |
| Pt_Ti/CPD_PED_2 | 4265.1 | 20.3 | 197.8 | 10.3 |
| Pt_Ti/CPD_PED_3 | 4825.8 | 23 | 219 | 10.5 |
| QDesorp/ µC cmgeo.−2 | SRF/ cmechem.² cmgeo.−2 | mPt/ µg cmgeo.−2 | mIr/ µg cmgeo.−2 | ECSA/ mechem.2 g−1 | |
|---|---|---|---|---|---|
| Pt + Ir_Ti/Two-Step | 10657.1 | 50.7 | 2470.3 | 5.7 | 2.1 |
| Tafel Parameter | ||||
|---|---|---|---|---|
| i@ 1.6 V vs. RHE/ mA cmgeo.−2 | MA/ A gPt + Ir−1 | b/ mV dek−1 | i0/ A m−² | |
| Pt + Ir_Ti/Two-Step | 9.885 | 5 | 428 | 14.1 |
| Ref. Ir-Disc | / | / | 68 | 1.1 × 10−4 |
| QDesorp/ µC cmgeo.−2 | SRF/ cmechem.2 cmgeo.−2 | mPt/ µg cmgeo.−2 | mIr/ µg cmgeo.−2 | ECSA/ mechem.2 g−1 | |
|---|---|---|---|---|---|
| Pt + Ir_Ti/One-Step | 74.9 | 0.4 | 12 | 15.2 | 1.3 |
| Tafel Parameter | |||||
|---|---|---|---|---|---|
| i@ 1.6 V vs. RHE/mA cmgeo.−2 | MA/ A gIr−1 | MA/ A gPt + Ir−1 | b/ mV dek−1 | i0/ A m−² | |
| Pt + Ir_Ti/One-Step | 0.523 | 34.4 | 19.2 | 436 | 1.32 |
| Ref. Ir-Disc | / | / | / | 68 | 1.1 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cieluch, M.; Podleschny, P.Y.; Kazamer, N.; Wirkert, F.J.; Rost, U.W.; Brodmann, M. Development of a Bifunctional Ti-Based Gas Diffusion Electrode for ORR and OER by One- and Two-Step Pt-Ir Electrodeposition. Nanomaterials 2022, 12, 1233. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12071233
Cieluch M, Podleschny PY, Kazamer N, Wirkert FJ, Rost UW, Brodmann M. Development of a Bifunctional Ti-Based Gas Diffusion Electrode for ORR and OER by One- and Two-Step Pt-Ir Electrodeposition. Nanomaterials. 2022; 12(7):1233. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12071233
Chicago/Turabian StyleCieluch, Maximilian, Pit Yannick Podleschny, Norbert Kazamer, Florian Josef Wirkert, Ulrich Wilhelm Rost, and Michael Brodmann. 2022. "Development of a Bifunctional Ti-Based Gas Diffusion Electrode for ORR and OER by One- and Two-Step Pt-Ir Electrodeposition" Nanomaterials 12, no. 7: 1233. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12071233