Nanostructured Dihydroartemisinin Plus Epirubicin Liposomes Enhance Treatment Efficacy of Breast Cancer by Inducing Autophagy and Apoptosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Liposomes
2.2. Cell Culture
2.3. Cellular Uptake and Mitochondrial Co-Localization
2.4. Induction of Autophagy
2.5. Induction of Apoptosis
2.6. Mechanisms of Autophagy and Apoptosis
2.7. Morphological Changes
2.8. Inhibitory Effects In Vitro
2.9. Anticancer Efficacy in Cancer-Bearing Mice
2.10. In Vivo Imaging
2.11. Statistical Analysis
3. Results
3.1. Characterization of Liposome Formulations
3.2. Cellular Uptake and Co-Localization Effect
3.3. Induction of Autophagy
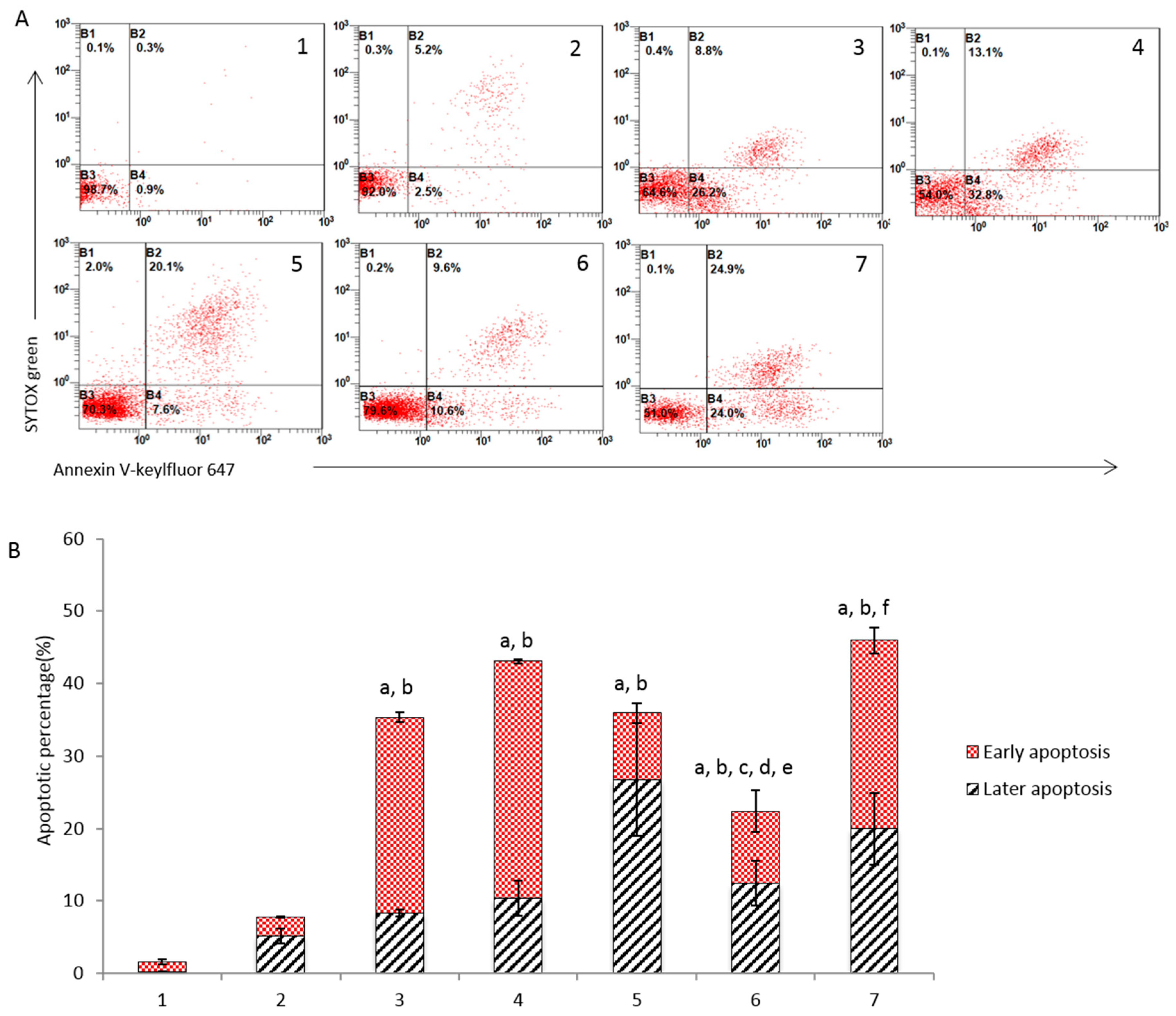
3.4. Induction of Apoptosis
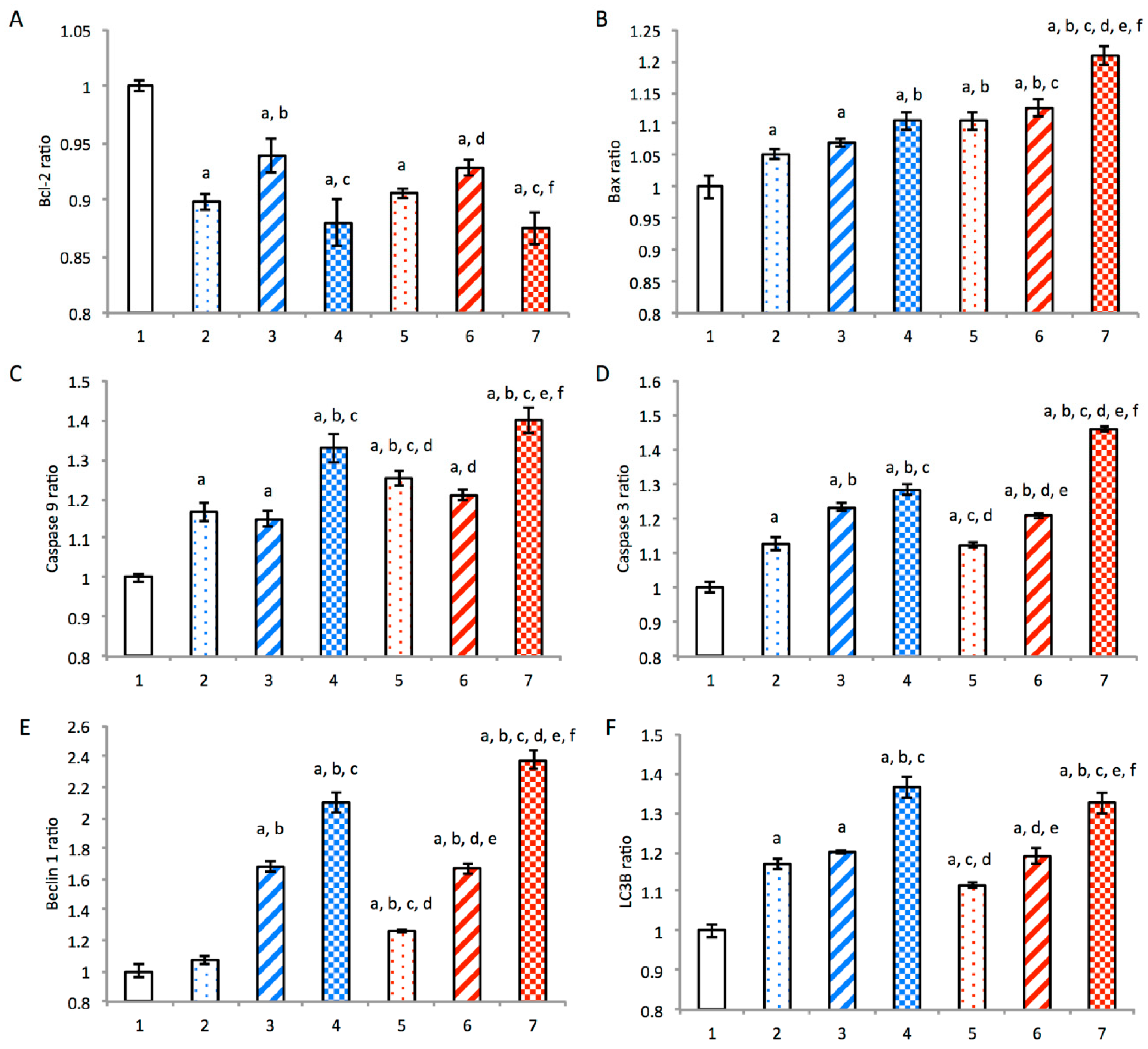
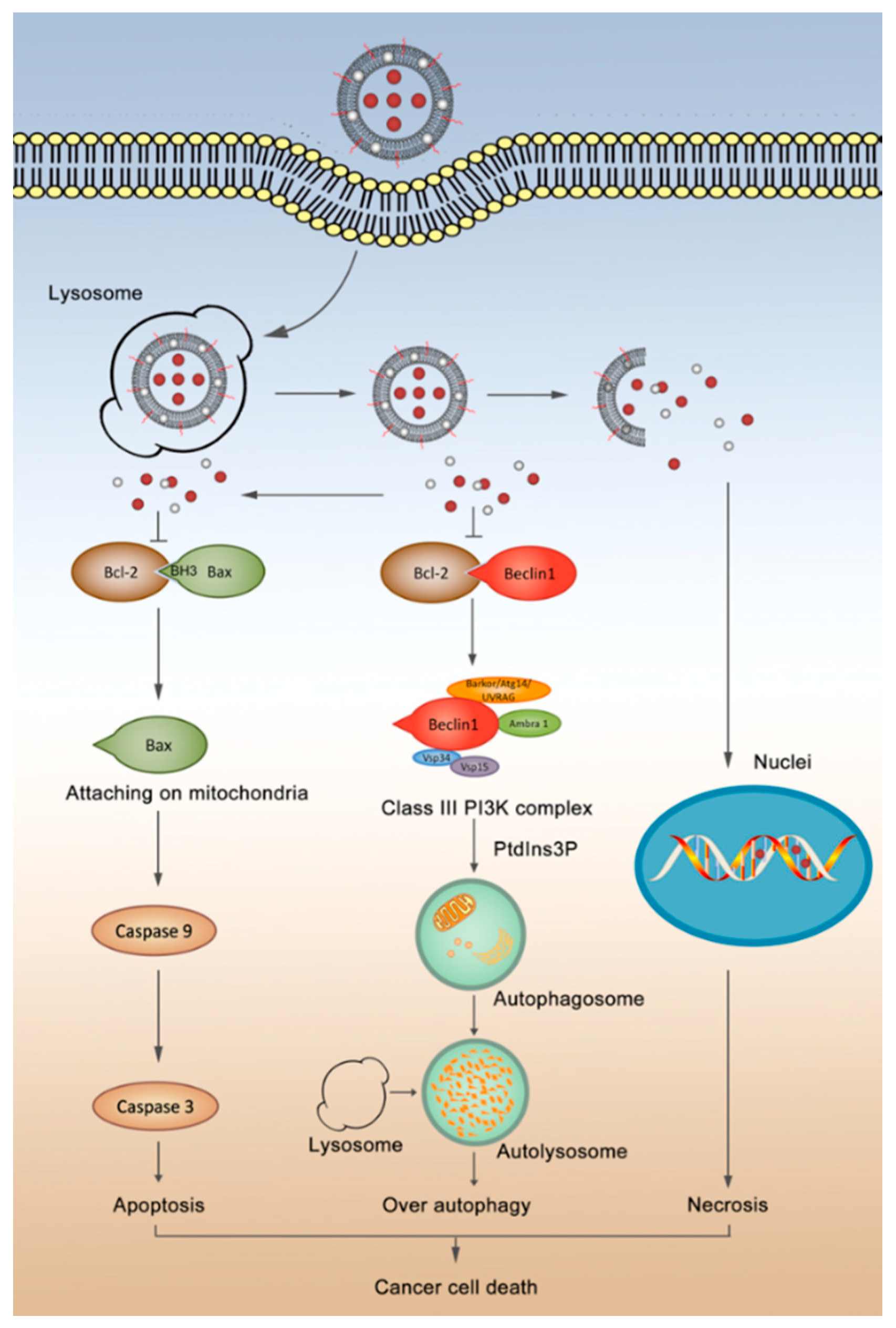
3.5. Mechanism of Autophagy and Apoptosis
3.6. Morphological Changes
3.7. Cytotoxic Effects In Vitro
3.8. Anticancer Efficacy and In Vivo Imaging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Rosenberg, P.S.; Prat, A.; Perou, C.M.; Sherman, M.E. How many etiological subtypes of breast cancer: Two, three, four, or more? J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24, S26–S35. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, R.; Perou, C.M.; Symmans, W.F.; Ibrahim, N.; Cristofanilli, M.; Anderson, K.; Hess, K.R.; Stec, J.; Ayers, M.; Wagner, P.; et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin. Cancer Res. 2005, 11, 5678–5685. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhang, X.Y.; Wang, Y.Q.; Zheng, X.L.; Zhao, Y.; Xing, W.M.; Zhang, Q. Andrographolide sodium bisulfate-induced apoptosis and autophagy in human proximal tubular endothelial cells is a ROS-mediated pathway. Environ. Toxicol. Pharmacol. 2014, 37, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Motyl, T.; Gajkowska, B.; Zarzynska, J.; Gajewska, M.; Lamparska-Przybysz, M. Apoptosis and autophagy in mammary gland remodeling and breast cancer chemotherapy. J. Physiol. Pharmacol. 2006, 57, 17–32. [Google Scholar] [PubMed]
- Zambrano, J.; Yeh, E.S. Autophagy and apoptotic crosstalk: Mechanism of therapeutic resistance in HER2-positive breast cancer. Breast Cancer 2016, 10, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS ONE 2009, 4, e6251. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Ahn, S.G.; Lee, B.H.; Jung, S.H.; Oh, S.H. Role of autophagy in chemoresistance: Regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem. Pharmacol. 2012, 83, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Garcia-Arencibia, M.; Zhao, R.; Puri, C.; Toh, P.P.C.; Sadiq, O.; Rubinsztein, D.C. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol. Cell 2012, 47, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y. The development of new antimalarial drugs: Qinghaosu and dihydro-qinghaosu. Chin. Med. J. 1999, 112, 976–977. [Google Scholar] [PubMed]
- Wu, C.; Liu, J.; Pan, X.; Xian, W.; Li, B.; Peng, W.; Wang, J.; Yang, D.; Zhou, H. Design, synthesis and evaluation of the antibacterial enhancement activities of amino dihydroartemisinin derivatives. Molecules 2013, 18, 6866–6882. [Google Scholar] [CrossRef] [PubMed]
- Flobinus, A.; Taudon, N.; Desbordes, M.; Labrosse, B.; Simon, F.; Mazeron, M.C.; Schnepf, N. Stability and antiviral activity against human cytomegalovirus of artemisinin derivatives. J. Antimicrob. Chemother. 2014, 69, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Aung, W.; Sogawa, C.; Furukawa, T.; Saga, T. Anticancer effect of dihydroartemisinin (DHA) in a pancreatic tumor model evaluated by conventional methods and optical imaging. Anticancer Res. 2011, 31, 1549–1558. [Google Scholar] [PubMed]
- Chen, H.H.; Zhou, H.J.; Fang, X. Inhibition of human cancer cell line growth and human umbilical vein endothelial cell angiogenesis by artemisinin derivatives in vitro. Pharmacol. Res 2003, 48, 231–236. [Google Scholar] [CrossRef]
- Jia, L.F.; Song, Q.; Zhou, C.Y.; Li, X.M.; Pi, L.H.; Ma, X.R.; Li, H.; Lu, X.; Shen, Y. Dihydroartemisinin as a putative STAT3 inhibitor, suppresses the growth of head and neck squamous cell carcinoma by targeting Jak2/STAT3 signaling. PLoS ONE 2016, 11, e0147157. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Gao, Y.; Chen, H.; Kong, R.; Jiang, H.C.; Pan, S.H.; Xue, D.B.; Bai, X.W.; Sun, B. Dihydroartemisinin inactivates NF-kB and potentiates the anti-tumor effect of gemcitabine on pancreatic cancer both in vitro and in vivo. Cancer Lett. 2010, 293, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Shi, L.; Ma, H.W.; Li, H.Z.; Li, Y.R.; Lu, Y.; Wang, Q.; Li, W. Dihydroartemisinin induces apoptosis in human gastric cancer cell line BGC-823 through activation of JNK1/2 and p38 MAPK signaling pathways. J. Recept. Signal Transduct. Res. 2016, 37, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Li, L.; Jiang, H.; Jiang, K.P.; Jin, Y.; Zheng, J.X. Dihydroartemisinin potentiates the anticancer effect of cisplatin via mTOR inhibition in cisplatin-resistant ovarian cancer cells: Involvement of apoptosis and autophagy. Biochem. Biophys. Res. Commun. 2014, 444, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Kong, R.; Ma, Z.B.; Han, B.; Wang, Y.W.; Pan, S.H.; Li, Y.H.; Sun, B. The activation of c-Jun NH2-terminal kinase is required for dihydroartemisinin-induced autophagy in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2014, 33, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.S.; Wang, J.; Shen, Y.B.; Guo, C.C.; Sai, K.; Chen, F.R.; Mei, X.; Han, F.; Chen, Z.P. Dihydroartemisinin increases temozolomide efficacy in glioma cells by inducing autophagy. Oncol. Lett. 2015, 10, 379–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Shou, L.M.; Lin, F.; Duan, W.M.; Wu, M.Y.; Xie, X.; Xie, Y.F.; Li, W.; Tao, M. Artesunate induces G2/M cell cycle arrest through autophagy induction in breast cancer cells. Anticancer Drugs 2014, 25, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Woodle, M.C.; Lasic, D.D. Sterically stabilized liposomes. Biochim. Biophys. Acta 1992, 1113, 171–199. [Google Scholar] [CrossRef]
- Maeda, H.; Tsukigawa, K.; Fang, J. A retrospective 30 years after discovery of the enhanced permeability and retention effect of solid tumors: Next-generation chemotherapeutics and photodynamic therapy--problems, solutions, and prospects. Microcirculation 2016, 23, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Ju, J.R.; Liu, L.; Xie, H.J.; Mu, L.M.; Zhao, Y.; Yan, Y.; Hu, Y.J.; Wu, J.S.; Lu, W.L. Application of functional vincristine plus dasatinib liposomes to deletion of vasculogenic mimicry channels in triple-negative breast cancer. Oncotarget 2015, 6, 36625–36642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, R.J.; Zeng, F.; Liu, L.; Mu, L.M.; Xie, H.J.; Zhao, Y.; Yan, Y.; Wu, J.S.; Hu, Y.J.; Lu, W.L. Destruction of vasculogenic mimicry channels by targeting epirubicin plus celecoxib liposomes in treatment of brain glioma. Int. J. Nanomed. 2016, 11, 1131–1146. [Google Scholar]
- Lu, W.L.; Qi, X.R.; Zhang, Q.; Li, R.Y.; Wang, G.L.; Zhang, R.J.; Wei, S.L. A pegylated liposomal platform: Pharmacokinetics, pharmacodynamics, and toxicity in mice using doxorubicin as a model drug. J. Pharmacol. Sci. 2004, 95, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Kogel, D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015, 34, 5105–5113. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Kabeya, Y.; Ohsumi, Y.; Yoshimori, T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001, 2, 330–335. [Google Scholar] [CrossRef] [PubMed]
- He, Y.Y.; Zhao, X.D.; Subahan, N.R.; Fan, L.F.; Gao, J.; Chen, H.L. The prognostic value of autophagy-related markers beclin-1 and microtubule-associated protein light chain 3B in cancers: A systematic review and meta-analysis. Tumour Biol. 2014, 35, 7317–7326. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. BioMed. Res. Int. 2014, 2014, 150845–150867. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Kim, D.; Kim, B.R.; Jun, J.S.; Yeom, J.S.; Park, J.S.; Seo, J.H.; Park, C.H.; Woo, H.O.; Youn, H.S.; et al. Vitamin C induces apoptosis in AGS cells via production of ROS of mitochondria. Oncol. Lett. 2016, 12, 4270–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karni, R.; Jove, R.; Levitzki, A. Inhibition of pp60c-Src reduces Bcl-XL expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene 1999, 18, 4654–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Klionsky, D.J. Physiological functions of Atg6/Beclin 1: A unique autophagy-related protein. Cell Res. 2007, 17, 839–849. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.-J.; Zhang, J.-Y.; Luo, Q.; Xu, J.-R.; Yan, Y.; Mu, L.-M.; Bai, J.; Lu, W.-L. Nanostructured Dihydroartemisinin Plus Epirubicin Liposomes Enhance Treatment Efficacy of Breast Cancer by Inducing Autophagy and Apoptosis. Nanomaterials 2018, 8, 804. https://0-doi-org.brum.beds.ac.uk/10.3390/nano8100804
Hu Y-J, Zhang J-Y, Luo Q, Xu J-R, Yan Y, Mu L-M, Bai J, Lu W-L. Nanostructured Dihydroartemisinin Plus Epirubicin Liposomes Enhance Treatment Efficacy of Breast Cancer by Inducing Autophagy and Apoptosis. Nanomaterials. 2018; 8(10):804. https://0-doi-org.brum.beds.ac.uk/10.3390/nano8100804
Chicago/Turabian StyleHu, Ying-Jie, Jing-Ying Zhang, Qian Luo, Jia-Rui Xu, Yan Yan, Li-Min Mu, Jing Bai, and Wan-Liang Lu. 2018. "Nanostructured Dihydroartemisinin Plus Epirubicin Liposomes Enhance Treatment Efficacy of Breast Cancer by Inducing Autophagy and Apoptosis" Nanomaterials 8, no. 10: 804. https://0-doi-org.brum.beds.ac.uk/10.3390/nano8100804