Effects of Sample Preparation on Particle Size Distributions of Different Types of Silica in Suspensions

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instruments and Procedures for Sample Preparation
2.3. Instruments for Particle Size Analysis
2.4. Estimation of the Calorimetric Energy Input
3. Results and Discussion
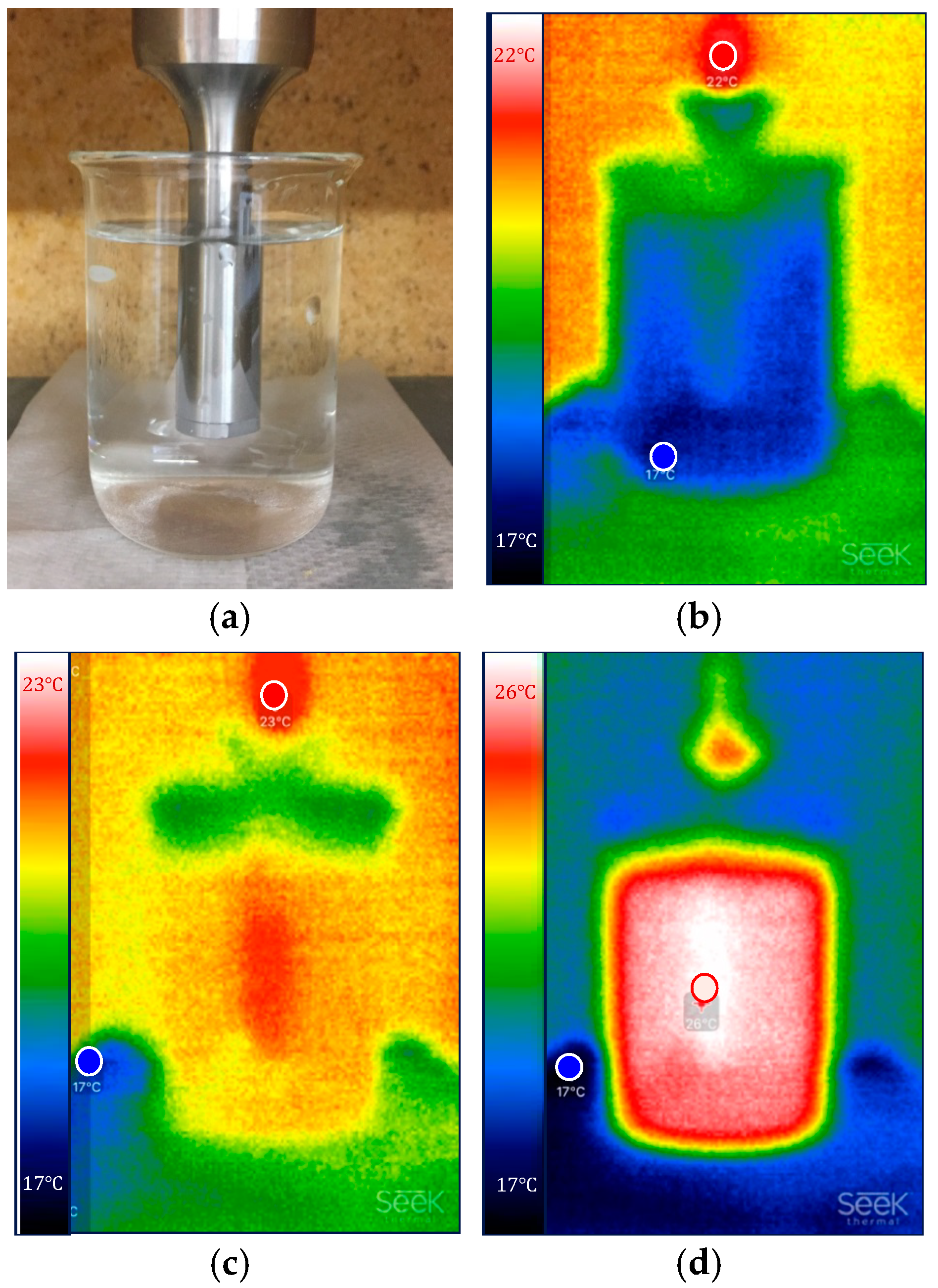
3.1. Calorimetric Calibration of Probe Sonication
3.2. Sample Preparation by Probe Sonication
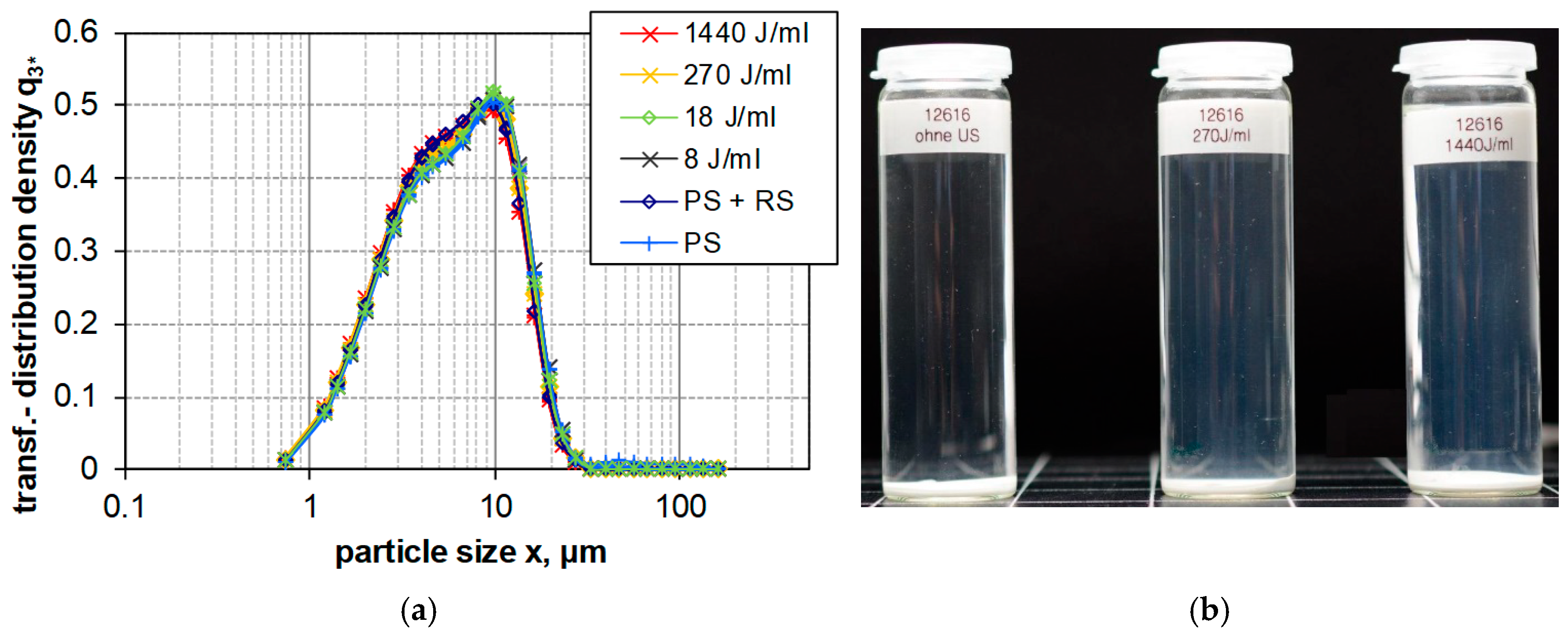
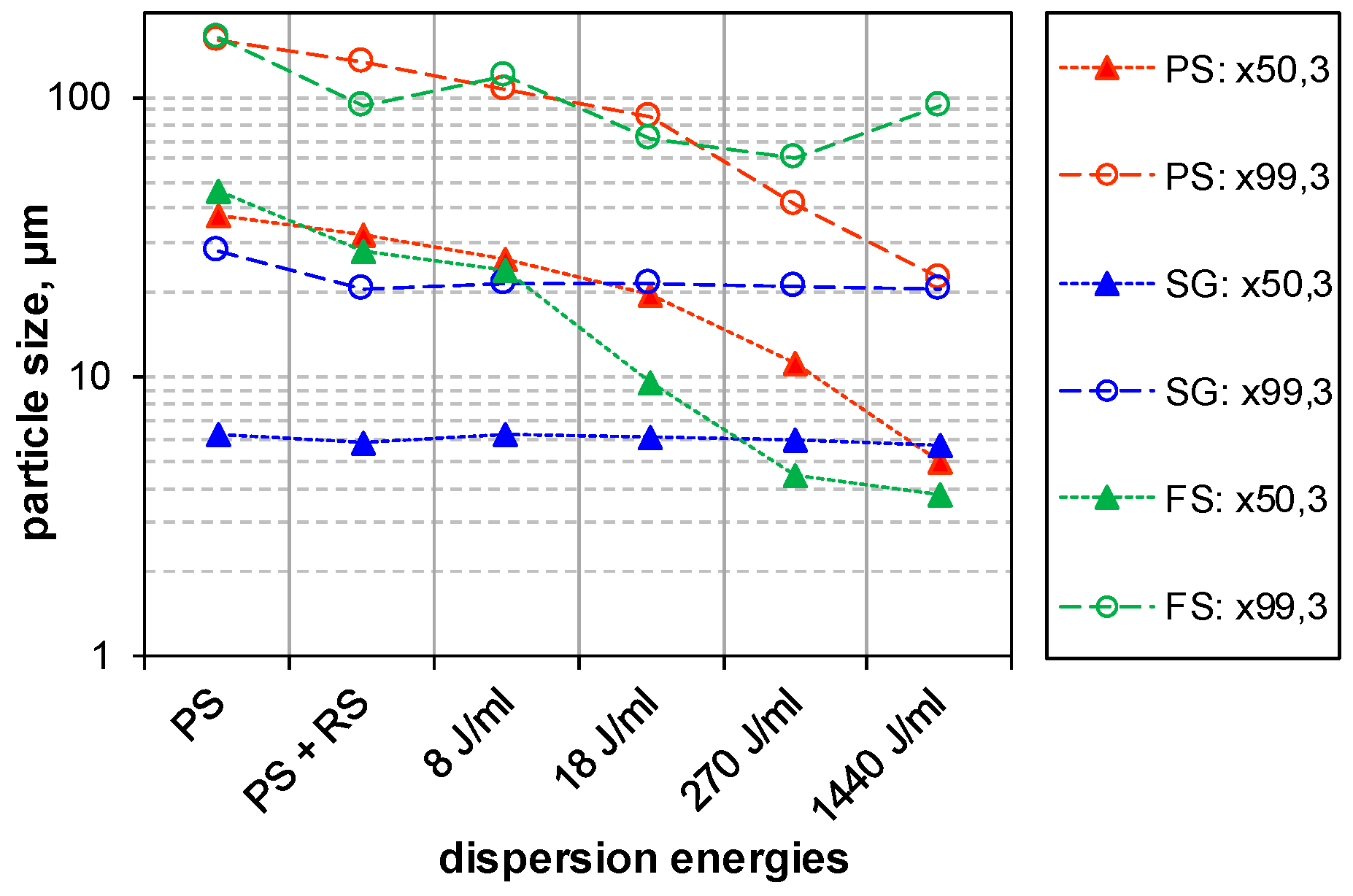
3.2.1. Impact of USD on Particle Size Distribution of SAS
3.2.2. Sample Contamination with Probe Sonication
3.3. Sample Preparation with Size-Selective Filtration
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borm, P.J.A.; Robbins, D.; Haubold, S.; Kuhlbusch, T.; Fissan, H.; Donaldson, K.; Schins, R.; Stone, V.; Kreyling, W.; Lademann, J.; et al. The potential risks of nanomaterials: A review carried out for ECETOC. Part. Fibre Toxicol. 2006, 3, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogharabi, M.; Abdollahi, M.; Faramarzi, M.A. Toxicity of nanomaterials; an undermined issue. DARU J. Pharm. Sci. 2014, 22, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruijtier-Pölloth, C. The safety of nanostructured synthetic amorphous silica (SAS) as a food additive (E 551). Arch. Toxicol. 2016, 90, 2885–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, C.; von Goetz, N.; Scheringer, M.; Wormuth, M.; Hungerbühler, K. Potential exposure of German consumers to engineered nanoparticles in cosmetics and personal care products. Nanotoxicology 2011, 5, 12–29. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Hwang, H.-M. Nanotechnology in food science: Functionality, applicability, and safety assessment. J. Food Drug Anal. 2016, 24, 671–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, H.C.; Suter, M.; Naegeli, H. Critical review of the safety assessment of nano-structured silica additives in food. J. Nanobiotechnol. 2016, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Retamal Marín, R.R.; Babick, F.; Stintz, M. Physico-chemical separation process of nanoparticles in cosmetic formulations. J. Phys. Conf. Ser. 2017, 838, 012004. [Google Scholar] [CrossRef] [Green Version]
- Froggett, S.J.; Clancy, S.F.; Boverhof, D.R.; Canady, R.A. A review and perspective of existing research on the release of nanomaterials from solid nanocomposites. Part. Fibre Toxicol. 2014, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Epstein, H.A.; Kielbassa, A. Nanotechnology in Cosmetic Products, Bio-Nanotechnology: A Revolution in Food. Biomed. Health Sci. 2013, 414–423. [Google Scholar] [CrossRef]
- Wu, M.S.; Sun, D.S.; Lin, Y.C.; Cheng, C.L.; Hung, S.C.; Chen, P.K.; Yang, J.H.; Chang, H.H. Nanodiamonds protect skin from ultraviolet B-induced damage in mice. J. Nanobiotechnol. 2015, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Huang, Y.W.; Zhou, X.D.; Ma, Y. In vitro toxicity of silica nanoparticles in human lung cancer cells. Toxicol. Appl. Pharmacol. 2006, 217, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Foged, C.; Brodin, B.; Frokjaer, S.; Sundblad, A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Noël, A.; Maghni, K.; Cloutier, Y.; Dion, C.; Wilkinson, K.J.; Hallé, S.; Tardif, R.; Truchon, G. Effects of inhaled nano-TiO2 aerosols showing two distinct agglomeration states on rat lungs. Toxicol. Lett. 2012, 214, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wu, Q.Y.; Li, M.Y.; Lao, C.S.; Zhang, Y.J. Pulmonary Toxicity in Rats Caused by Exposure to Intratracheal Instillation of SiO2 Nanoparticles. Biomed. Environ. Sci. 2017, 30, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, M.; Vennemann, A.; Sauer, U.G.; Wiench, K.; Ma-Hock, L.; Landsiedel, R. An in vitro alveolar macrophage assay for predicting the short-term inhalation toxicity of nanomaterials. J. Nanobiotechnol. 2016, 14, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marvanová, S.; Kulich, P.; Skoupý, R.; Hubatka, F.; Ciganek, M. Size-segregated urban aerosol characterization by electron microscopy and dynamic light scattering and influence of sample preparation. Atmos. Environ. 2018. [Google Scholar] [CrossRef]
- Benelli, G. Mode of action of nanoparticles against insects. Environ. Sci. Pollut. Res. 2018, 25, 12329–12341. [Google Scholar] [CrossRef] [PubMed]
- Babick, F. Suspensions of colloidal particles and aggregates. In Particle Technology Series; Valverde Millán, J.M., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; Volume 20, ISBN 978-3-319-30661-2. [Google Scholar]
- Babick, F.; Schieß, K.; Stintz, M. Characterization of Pyrogenic Powders with Conventional Particle Sizing Technique: I. Prediction of Measured Size Distributions. Part. Part. Syst. Charact. 2012, 29, 104–115. [Google Scholar] [CrossRef]
- Retamal Marín, R.R.; Babick, F.; Stintz, M. Ultrasonic dispersion of nanostructured materials with probe sonication—Practical aspects of sample preparation. Powder Technol. 2017, 318, 451–458. [Google Scholar] [CrossRef]
- Oberdörster, G.; Maynard, A.; Donaldson, K.; Castranova, V.; Fitzpatrick, J.; Ausman, K.; Carter, J.; Karn, B.; Kreyling, W.; Lai, D.; et al. Principles for characterizing the potential human health effects from exposure to nanomaterials: Elements of a screening strategy. Part. Fibre Toxicol. 2005, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Passagne, I.; Morille, M.; Rousset, M.; Pujalté, I.; L’Azou, B. Implication of oxidative stress in size-dependent toxicity of silica nanoparticles in kidney cells. Toxicology 2012, 299, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Taurozzi, J.; Hackley, V.; Wiesner, M. Ultrasonic dispersion of nanoparticles for environmental, health and safety assessment—Issues and recommendations. Nanotoxicology 2011, 5, 711–729. [Google Scholar] [CrossRef] [PubMed]
- Veith, L.; Vennemann, A.; Breitenstein, D.; Engelhard, C.; Wiemann, M.; Hagenhoff, B. Detection of SiO2 nanoparticles in lung tissue by ToF-SIMS imaging and fluorescence microscopy. Analyst 2017, 142, 2631. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.; Hannebauer, B.; Holldorff, H.; Albers, P. Does Lung Surfactant Promote Disaggregation of Nanostructured Titanium Dioxide? J. Occup. Environ. Med. 2006, 48, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Bakand, S.; Hayes, A.; Dechsakulthorn, F. Nanoparticles: A review of particle toxicology following inhalation exposure. Inhal. Toxicol. 2012, 24, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Ahluwalia, A.; Allen, D.; Bonner, J.C.; Casey, W.; Castranova, V.; David, R.M.; Halappanavar, S.; Hotchkiss, J.A.; Jarabek, A.M.; et al. Expert consensus on an in vitro approach to assess pulmonary fibrogenic potential of aerosolized nanomaterials. Arch. Toxicol. 2016, 90, 1769–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemmar, A.; Hoylaerts, M.F.; Hoet, P.H.M.; Vermylen, J.; Nemery, B. Size effect of intratracheally instilled particles on pulmonary inflammation and vascular thrombosis. Toxicol. Appl. Pharmacol. 2003, 186, 38–45. [Google Scholar] [CrossRef]
- Arick, D.Q.; Choi, Y.H.; Kim, H.C.; Won, Y. Effects of nanoparticles on the mechanical functioning of the lung. Adv. Colloid Interface Sci. 2015, 225, 218–228. [Google Scholar] [CrossRef] [PubMed]
- ISO. Nanotechnologies—Health and Safety Practices in Occupational Settings Relevant to Nanotechnologies; TC229, ISO/TR 12885; ISO: Geneva, Switzerland, 2008. [Google Scholar]
- European Commission. Commission Recommendation of 18 October 2011 on the Definition of Nanomaterial (2011/696/EU). Off. J. Eur. Union 2011, 54, 38–40. [Google Scholar]
- Linsinger, T.P.J.; Roebben, G.; Gilliland, D.; Calzolai, L.; Rossi, F.; Gibson, N.; Klein, C. Requirements on Measurements for the Implementation of the European Commission Definition of the Term ‘Nanomaterial’; Publications Office of the European Union: Luxembourg, 2012. [Google Scholar] [CrossRef]
- Jensen, K.; Kembouche, Y.; Christiansen, E.; Jacobsen, N.; Wallin, H.; Guiot, C.; Spalla, O.; Witschger, O. The Generic NANOGENOTOX Dispersion Protocol: Final Protocol for Producing Suitable Manufactured Nanomaterial Exposure Media. NANOGENOTOX Joint Action, European Commission. 2011. Available online: www.nanogenotox.eu/files/PDF/web%20nanogenotox%20dispersion%20protocol.pdf (accessed on 20 June 2018).
- OECD. Guidelines for the Testing of Chemicals, Section 3 Test No. 318: Dispersion Stability of Nanomaterials in Simulated Environmental Media; OECD Publishing: Washington, DC, USA, 2017; ISBN 9789264284142. [Google Scholar]
- Rasmussen, K.; Mech, A.; Mast, J.; de Temmerman, P.-J.; Waegeneers, N.; van Steen, F.; Pizzolon, J.C.; de Temmerman, L.; van Doren, E.; Jensen, K.A.; et al. Synthetic Amorphous Silicon Dioxide (NM-200, NM-201, NM-202, NM-203, NM-204): Characterisation and Physico-Chemical Properties; Report EUR 26046; European Commission: Brussels, Belgium, 2013. [Google Scholar]
- Hartmann, N.; Jensen, K.; Baun, A.; Rasmussen, K.; Rauscher, H.; Tantra, R.; Cupi, D.; Gilliland, D.; Pianella, F.; Sintes, J.R. Techniques and Protocols for Dispersing Nanoparticle Powders in Aqueous Media—Is there a Rationale for Harmonization? J. Toxicol. Environ. Health B 2015, 18, 299–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, M.; Schubert, H.; Schuchmann, H. Herstellung stabiler Dispersionen aus pyrogener Kieselsäure. Chem. Ing. Tech. 2005, 77, 258–262. [Google Scholar] [CrossRef]
- Wengeler, R.; Ruslim, F.; Nirschl, H.; Merkel, T. Dispergierung feindisperser Agglomerate mit Mikro-Dispergierelementen. Chem. Ing. Tech. 2004, 76, 659–662. [Google Scholar] [CrossRef]
- Wengeler, R.; Teleki, A.; Vetter, M.; Pratsinis, S.; Nirschl, H. High-pressure liquid dispersion and fragmentation of flame-made silica agglomerates. Langmuir 2006, 22, 4928–4935. [Google Scholar] [CrossRef] [PubMed]
- Sauter, C.; Schuchmann, H. High pressure for dispersing and deagglomerating nanoparticles in aqueous solutions. Chem. Eng. Technol. 2007, 30, 1401–1405. [Google Scholar] [CrossRef]
- Bałdyga, J.; Makowski, Ł.; Orciuch, W.; Sauter, C.; Schuchmann, H. Agglomerate dispersion in cavitating flows. Chem. Eng. Res. Des. 2009, 87, 474–484. [Google Scholar] [CrossRef]
- Tantra, R. Nanomaterial Characterization: An Introduction; Wiley: Hoboken, NJ, USA, 2016; ISBN 9781118753460. [Google Scholar]
- Pradhan, S.; Hedberg, J.; Wold, E.B.S.; Wallinder, I.O. Effect of sonication on particle dispersion, administered dose and metal release of non-functionalized, non-inert metal nanoparticles. J. Nanopart Res. 2016, 18, 285. [Google Scholar] [CrossRef] [PubMed]
- Taurozzi, J.; Hackley, V.; Wiesner, M. Preparation of Nanoparticle Dispersions from Powdered Material using Ultrasonic Disruption. NIST Spec. Publ. 2012. [Google Scholar] [CrossRef]
- Bihari, P.; Vippola, M.; Schultes, S.; Praetner, M.; Khandoga, A.G.; Reichel1, C.A.; Coester, C.; Tuomi, T.; Rehberg, M.; Krombach, F. Optimized dispersion of nanoparticles for biological in vitro and in vivo studies. Part. Fibre Toxicol. 2008, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandzy, N.; Grulke, E.; Druffel, T. Breakage of TiO2 agglomerates in electrostatically stabilized aqueous dispersions. Powder Technol. 2005, 160, 121–126. [Google Scholar] [CrossRef]
- Pohl, M.; Hogekamp, S.; Hoffmann, N.; Schuchmann, H. Dispergieren und Desagglomerieren von Nanopartikeln mit Ultraschall. Chem. Ing. Tech. 2004, 76, 392–396. [Google Scholar] [CrossRef]
- Napierska, D.; Thomassen, L.C.J.; Lison, D.; Martens, J.A.; Hoet, P.H. The nanosilica hazard: Another variable entity. Part. Fibre Toxicol. 2010, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinker, C.F.; Schrerer, G.W. Sol-Gel Science. The Physics and Chemistry of Sol-Gel Processing, 2nd ed.; Academic Press: London, UK, 1990; ISBN 9780121349707. [Google Scholar]
- Albers, P.; Maier, M.; Reisinger, M.; Hannebauer, B.; Weinand, R. Physical boundaries within aggregates—differences between amorphous, para-crystalline, and crystalline Structures. Cryst. Res. Technol. 2015, 50, 846–865. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 309–319. [Google Scholar] [CrossRef]
- ISO 9277:2010. Determination of the Specific Surface Area of Solids by Gas Adsorption—BET Method; ISO: Geneva, Switzerland, 2010. [Google Scholar]
- Hosokawa, M.; Nogi, K.; Naito, M.; Yokoyama, T. Nanoparticle Technology Handbook; Elsevier Science: New York, NY, USA, 2007; ISBN 978-0-444-53122-3. [Google Scholar]
- Retamal Marín, R.R.; Babick, F.; Hillemann, L. Zeta potential measurements for non-spherical colloidal particles—Practical issues of characterisation of interfacial properties of nanoparticles. Colloids Surf. A 2017, 532, 516–521. [Google Scholar] [CrossRef]
- Karina Maria Paciejewska, Untersuchung des Stabilitätsverhaltens von binären kolloidalen Suspensionen, Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany, December 2010. Available online: http://d-nb.info/1019001267/34 (accessed on 20 June 2018).
- European Pharmacopoeia: Supplement 5.7, Band 5 von European Pharmacopoeia: Supplement, 5th ed.; Convention on the Elaboration of a European Pharmacopoeia, 4805–4806; Council of Europe: Strasbourg, France, 2006.
- Hauptmann, P.; Sorge, G. Ultraschall in Wissenschaft und Technik; Durchschnittliche Kundenbewertung: Leipzig, Germany, 1985; ISBN 0-906674-38-7. [Google Scholar]
- Kusters, K.; Pratsinis, S.; Thoma, S.; Smith, D. Ultrasonic fragmentation of agglomerate powders. Chem. Eng. Sci. 1993, 48, 4119–4127. [Google Scholar] [CrossRef]
- Aoki, M.; Ring, T.; Haggerty, J. Analysis and modeling of the ultrasonic dispersion technique. Adv. Ceram. Mater. 1987, 2, 209–212. [Google Scholar] [CrossRef]
- Etzler, F.M.; Deanne, R. Particle Size Analysis: A Comparison of Various Methods II. Part. Part. Syst. Charact. 1997, 14, 278–282. [Google Scholar] [CrossRef]
- Bayat, H.; Rastgo, M.; Zadeh, M.M.; Vereecken, H. Particle size distribution models, their characteristics and fitting capability. J. Hydrol. 2015, 529, 872–889. [Google Scholar] [CrossRef]
- Marton, L.; Marton, C. Methods of Experimental Physics: Ultrasonics; Academic Press: Cambridge, MA, USA, 1981; ISBN 0-12-475961-0. [Google Scholar]
- Raman, V.; Abbas, A. Experimental investigations on ultrasound mediated particle breakage. Ultrason. Sonochem. 2008, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Raso, J.; Mañas, P.; Pagán, R.; Sala, F. Influence of different factors on the output power transferred into medium by ultrasound. Ultrason. Sonochem. 1999, 5, 157–162. [Google Scholar] [CrossRef]
- Edmond, P.D. Methods in Experimental Physics; Elsevier: New York, NY, USA, 1981; Volume 19, ISBN 978-0-12-475961-9. [Google Scholar]
- Bhatia, A.B. Ultrasonic Absorption. An Introduction to the Theory of Sound Absorption and Dispersion in Gases, Liquids, and Solids; Oxford University Press: New York, NY, USA, 1967; ISBN 9780486649177. [Google Scholar]
- ISO 9276-1:1998. Representation of Results of Particle size Analysis—Part 1: Graphical Representation; ISO: Geneva, Switzerland, 1998. [Google Scholar]
- Kuchenbecker, P.; Gemeinert, M.; Rabe, T. Interlaboratory study of particle size distribution measurements by laser diffraction. Part. Part. Syst. Charact. 2012, 29, 304–310. [Google Scholar] [CrossRef]
- Mawson, R.; Rout, M.; Ripoll, G.; Swiergon, P.; Singh, T.; Koerzer, K.; Juliano, P. Production of particulates from transducer erosion: Implications on food safety. Ultrason. Sonochem. 2014, 21, 2122–2130. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protocol | Sample Volume | Dispersing Time | (Calorimetric) Energy Density |
|---|---|---|---|
| Tantra 2016 [42]; Pradhan 2016 [43] | 6 mL | 16 min | 1176 J/mL |
| Rasmussen et al., 2013 [35] | 15 mL 10 mL | 10 min 16 min | 500–400 J/mL 2500 J/mL |
| Taurozzi et al., 2012 [44] | 50 mL | 5 min | 300 J/mL |
| Jensen et al., 2011 [33] | 6 mL | 16 min | 3140 J/mL |
| Bihari et al., 2008 [45] | 1 mL | 1 min | 420 J/mL |
| Mandzy et al., 2005 [46] | - | Time frames (2 h) | 5700 J/mL |
| Pohl et al., 2005 [37] | 10–42 mL | 17–630 s | 400–30,000 J/mL |
| Pohl et al., 2004 [47] | 3–6 mL | - | 100–2000 J/mL |
| SAS Type Internal Code | Fumed Silica F-3 | Precipitated Silica P-2 | Silica Gel G-1 | Colloidal Silica C-1 |
|---|---|---|---|---|
| BET 1 (m2/g) | 300 | 440 | 700 | 200 2 |
| solid content for suspensions (wt.-%) | - | - | - | 40 |
| pH 3 | 5 | 6.5 | 4.4 | 9.7 |
| electric conductivity (μS/cm) at 25 °C | 4 | 160 | 55 | 4771.6 |
| Model | Vibra-Cell 72412 1 | UDS751 2 | SONIFIER 450D 3 |
|---|---|---|---|
| Code | V | T | B |
| company | Sonics and Materials | Topas GmbH | Branson Ultrasonics |
| normal capacity (W) | 600 | 200 | 400 |
| tip diameter (Ø, mm) | 13 19 | 3 7 14 | 5 13 |
| amplitude (%) | 0–100 | 0–100 | 10–100 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Retamal Marín, R.R.; Babick, F.; Lindner, G.-G.; Wiemann, M.; Stintz, M. Effects of Sample Preparation on Particle Size Distributions of Different Types of Silica in Suspensions. Nanomaterials 2018, 8, 454. https://0-doi-org.brum.beds.ac.uk/10.3390/nano8070454
Retamal Marín RR, Babick F, Lindner G-G, Wiemann M, Stintz M. Effects of Sample Preparation on Particle Size Distributions of Different Types of Silica in Suspensions. Nanomaterials. 2018; 8(7):454. https://0-doi-org.brum.beds.ac.uk/10.3390/nano8070454
Chicago/Turabian StyleRetamal Marín, Rodrigo R., Frank Babick, Gottlieb-Georg Lindner, Martin Wiemann, and Michael Stintz. 2018. "Effects of Sample Preparation on Particle Size Distributions of Different Types of Silica in Suspensions" Nanomaterials 8, no. 7: 454. https://0-doi-org.brum.beds.ac.uk/10.3390/nano8070454