Electrochemical Detection of Ultratrace Lead Ion through Attaching and Detaching DNA Aptamer from Electrochemically Reduced Graphene Oxide Electrode


Abstract
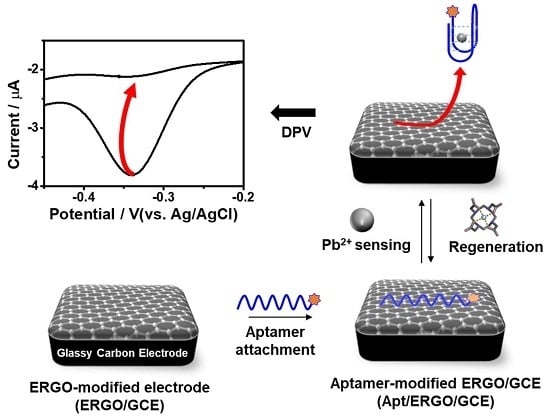
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrumentation
2.3. Fabrication of Apt/ERGO/GCE Sensor
3. Results and Discussion
3.1. Charaterization of Apt/ERGO/GCE-Based Pb2+ Sensor
3.2. Optimization of Experimental Conditions
3.3. Electroanalytical Performance of Apt/ERGO/GCE-Based Pb2+ Sensor
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Claudio, E.S.; Goldwin, H.A.; Magyar, J.S. Fundamental coordination chemistry, environmental chemistry, and biochemistry of lead (II). Prog. Inorg. Chem. 2003, 51, 1–144. [Google Scholar] [CrossRef]
- Needleman, H. Lead poisoning. Annu. Rev. Med. 2004, 55, 209–222. [Google Scholar] [CrossRef]
- Godwin, H.A. The biological chemistry of lead. Curr. Opin. Chem. Biol. 2001, 5, 223–227. [Google Scholar] [CrossRef]
- Butler, O.T.; Cook, J.M.; Harrington, C.F.; Hill, S.J.; Rieuwerts, J.; Miles, D.L. Atomic spectrometry update. Environmental analysis. J. Anal. At. Spectrom. 2007, 22, 187–221. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, H.; Mao, X.; Jin, X. Determination of trace copper, lead, cadmium, and iron in environmental and biological samples by flame atomic absorption spectrometry coupled to flow injection on-line coprecipitation preconcentration using DDTC-nickel as coprecipitate carrier. Int. J. Environ. Anal. Chem. 2000, 76, 267–282. [Google Scholar] [CrossRef]
- Ochsenkühn-Petropoulou, M.; Ochsenkühn, K.-M. Comparison of inductively coupled plasma–atomic emission spectrometry, anodic stripping voltammetry and instrumental neutron-activation analysis for the determination of heavy metals in airborne particulate matter. Fresenius J. Anal. Chem. 2001, 369, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Ettler, V.; Mihaljevič, M.; Komárek, M. ICP-MS measurements of lead isotopic ratios in soils heavily contaminated by lead smelting: Tracing the sources of pollution. Anal. Bioanal. Chem. 2004, 378, 311–317. [Google Scholar] [CrossRef]
- Yang, D.; Wang, L.; Chen, Z.; Megharaj, M.; Naidu, R. Anodic stripping voltammetric determination of traces of Pb(II) and Cd(II) using a glassy carbon electrode modified with bismuth nanoparticles. Microchim. Acta 2014, 181, 1199–1206. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, X.; Niyungeko, C.; Zhou, J.; Xu, J.; Tian, G. A review of the identification and detection of heavy metal ions in the environment by voltammetry. Talanta 2018, 178, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, M.A.; Shirsat, M.D.; Ramanaviciene, A.; Ramanavicius, A. Composites Based on Conducting Polymers and Carbon Nanomaterials for Heavy Metal Ion Sensing (Review). Crit. Rev. Anal. Chem. 2018, 48, 293–304. [Google Scholar] [CrossRef]
- Mishra, G.K.; Sharma, V.; Mishra, R.K. Electrochemical Aptasensors for Food and Environmental Safeguarding: A Review. Biosensors 2018, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, X.; Zhou, Y.; Luo, L.; Zhang, J.; Huang, A.; Mao, Q.; Chen, X.; Tang, L. Aptamer-based biosensors for detection of lead ion: A review. Anal. Methods 2017, 9, 1976–1990. [Google Scholar] [CrossRef]
- Qing, M.; Yuan, Y.; Cai, W.; Xie, S.; Tang, Y.; Yuan, R.; Zhang, J. An ultrasensitive electrochemical biosensor based on multifunctional hemin/G-quadruplex nanowires simultaneously served as bienzyme and direct electron mediator for detection of lead ion. Sens. Actuators B Chem. 2018, 263, 469–475. [Google Scholar] [CrossRef]
- Xiao, Y.; Rowe, A.A.; Plaxco, K.W. Electrochemical Detection of Parts-Per-Billion Lead via an Electrode-Bound DNAzyme Assembly. J. Am. Chem. Soc. 2007, 129, 262–263. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tang, L.; Zeng, G.; Zhang, C.; Zhang, Y.; Xie, X. Current progress in biosensors for heavy metal ions based on DNAzymes/DNA molecules functionalized nanostructures: A review. Sens. Actuators B Chem. 2016, 223, 280–294. [Google Scholar] [CrossRef]
- Pumera, M. Electrochemistry of graphene, graphene oxide and other graphenoids: Review. Electrochem. Commun. 2013, 36, 14–18. [Google Scholar] [CrossRef]
- Chen, D.; Feng, H.; Li, J. Graphene Oxide: Preparation, Functionalization, and Electrochemical Applications. Chem. Rev. 2012, 112, 6027–6053. [Google Scholar] [CrossRef] [PubMed]
- Toh, S.Y.; Loh, K.S.; Kamarudin, S.K.; Daud, W.R.W. Graphene production via electrochemical reduction of graphene oxide: Synthesis and characterisation. Chem. Eng. J. 2014, 251, 422–434. [Google Scholar] [CrossRef]
- Sham, A.Y.W.; Notley, S.M. A review of fundamental properties and applications of polymer–graphene hybrid materials. Soft Matter 2013, 9, 6645–6653. [Google Scholar] [CrossRef]
- Shahriary, L.; Athawale, A.A. Graphene oxide synthesized by using modified hummers approach. Int. J. Renew. Energy Environ. Eng. 2014, 2, 58–63. [Google Scholar]
- Lee, C.-S.; Yu, S.H.; Kim, T.H. One-Step Electrochemical Fabrication of Reduced Graphene Oxide/Gold Nanoparticles Nanocomposite-Modified Electrode for Simultaneous Detection of Dopamine, Ascorbic Acid, and Uric Acid. Nanomaterials 2017, 8, 17. [Google Scholar] [CrossRef]
- Yu, J.; Kim, T.H. A Facile Electrochemical Fabrication of Reduced Graphene Oxide-Modified Glassy Carbon Electrode for Simultaneous Detection of Dopamine, Ascorbic Acid, and Uric Acid. J. Electrochem. Sci. Technol. 2017, 8, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Tang, Y.; Wang, K.; Liu, C.; Luo, S. Direct electrodeposition of reduced graphene oxide on glassy carbon electrode and its electrochemical application. Electrochem. Commun. 2011, 13, 133–137. [Google Scholar] [CrossRef]
- Li, T.; Wang, E.; Dong, S. Lead(II)-Induced Allosteric G-Quadruplex DNAzyme as a Colorimetric and Chemiluminescence Sensor for Highly Sensitive and Selective Pb2+ Detection. Anal. Chem. 2010, 82, 1515–1520. [Google Scholar] [CrossRef]
- Shao, Y.; Wang, J.; Engelhard, M.; Wang, C.; Lin, Y. Facile and controllable electrochemical reduction of graphene oxide and its applications. J. Mater. Chem. 2010, 20, 743–748. [Google Scholar] [CrossRef]
- Yang, D.; Velamakanni, A.; Bozoklu, G.; Park, S.; Stoller, M.; Piner, R.D.; Stankovich, S.; Jung, I.; Field, D.A.; Ventrice, C.A.; et al. Chemical analysis of graphene oxide films after heat and chemical treatments by X-ray photoelectron and Micro-Raman spectroscopy. Carbon 2009, 47, 145–152. [Google Scholar] [CrossRef]
- Lee, H.-K.; Heo, J.; Myung, S.; Shin, I.-S.; Kim, T.H. Homogeneous Electrochemical Assay for Real-time Monitoring of Exonuclease III Activity Based on a Graphene Monolayer. Electroanalysis 2017, 29, 1749–1754. [Google Scholar] [CrossRef]
- Satapathy, D.R.; Salve, P.R.; Katpatal, Y.B. Spatial distribution of metals in ground/surface waters in the Chandrapur district (Central India) and their plausible sources. Environ. Geol. 2009, 56, 1323–1352. [Google Scholar] [CrossRef]
- Cheng, M.-T.; Chio, C.-P.; Huang, C.-Y.; Chen, J.-M.; Wang, C.-F.; Kuo, C.-Y. Chemical compositions of fine particulates emitted from oil-fired boilers. J. Environ. Eng. Manag. 2008, 18, 355–362. [Google Scholar]
- Liang, L.; Lan, F.; Ge, S.; Yu, J.; Ren, N.; Yan, M. Metal-Enhanced Ratiometric Fluorescence/Naked Eye Bimodal Biosensor for Lead Ions Analysis with Bifunctional Nanocomposite Probes. Anal. Chem. 2017, 89, 3597–3605. [Google Scholar] [CrossRef]
- Yun, W.; Cai, D.; Jiang, J.; Zhao, P.; Huang, Y.; Sang, G. Enzyme-free and label-free ultra-sensitive colorimetric detection of Pb2+ using molecular beacon and DNAzyme based amplification strategy. Biosens. Bioelectron. 2016, 80, 187–193. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, H.; Jiang, X.; Sun, B.; Song, B.; Su, Y.; He, Y. Ultrasensitive, Specific, Recyclable, and Reproducible Detection of Lead Ions in Real Systems through a Polyadenine-Assisted, Surface-Enhanced Raman Scattering Silicon Chip. Anal. Chem. 2016, 88, 3723–3729. [Google Scholar] [CrossRef]
- Cui, L.; Wu, J.; Li, J.; Ju, H. Electrochemical Sensor for Lead Cation Sensitized with a DNA Functionalized Porphyrinic Metal–Organic Framework. Anal. Chem. 2015, 87, 10635–10641. [Google Scholar] [CrossRef]
- Pelossof, G.; Tel-Vered, R.; Willner, I. Amplified Surface Plasmon Resonance and Electrochemical Detection of Pb2+ Ions Using the Pb2+-Dependent DNAzyme and Hemin/G-Quadruplex as a Label. Anal. Chem. 2012, 84, 3703–3709. [Google Scholar] [CrossRef]
- Zhou, Y.; Tang, L.; Zeng, G.; Zhang, C.; Xie, X.; Liu, Y.; Wang, J.; Tang, J.; Zhang, Y.; Deng, Y. Label free detection of lead using impedimetric sensor based on ordered mesoporous carbon–gold nanoparticles and DNAzyme catalytic beacons. Talanta 2016, 146, 641–647. [Google Scholar] [CrossRef]
- Zhao, J.; Jing, P.; Xue, S.; Xu, W. Dendritic structure DNA for specific metal ion biosensor based on catalytic hairpin assembly and a sensitive synergistic amplification strategy. Biosens. Bioelectron. 2017, 87, 157–163. [Google Scholar] [CrossRef]
- Wang, L.; Wen, Y.; Li, L.; Yang, X.; Jia, N.; Li, W.; Meng, J.; Duan, M.; Sun, X.; Liu, G. Sensitive and label-free electrochemical lead ion biosensor based on a DNAzyme triggered G-quadruplex/hemin conformation. Biosens. Bioelectron. 2018, 115, 91–96. [Google Scholar] [CrossRef]
- Viola-Villegas, N.; Doyle, R.P. The coordination chemistry of 1,4,7,10-tetraazacyclododecane-N,N′,N″,N′″-tetraacetic acid (H4DOTA): Structural overview and analyses on structure–stability relationships. Coord. Chem. Rev. 2009, 253, 1906–1925. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Detection Limit | Linear Range (M) | Ref. |
|---|---|---|---|
| ICP-MS | 0.38 nM | - | [28] |
| ICP-AES | 40 nM | - | [29] |
| Fluorescence | 0.3 pM | 1.0 × 10−12–3.0 × 10−6 | [30] |
| Colorimetry | 20 pM | 5 ×10−11–5 × 10−9 | [31] |
| SERS | 8.9 pM | 10−11–10−6 | [32] |
| ASV | 3.9 nM | 2.4 × 10−10–2.9 × 10−7 | [8] |
| DPV | 0.51 fM | 10−15–10−9 | This work |
| Electrode Materials | Detection Limit | Linear Range (M) | Ref. |
| DNAzyme/Au | 300 nM | 5 × 10−7–10−5 | [14] |
| HP/AuNPs/SPCE | 34 pM | 5 × 10−11–2 × 10−7 | [33] |
| DNAzyme/AuNP/Au | 100 pM | 10−10–10−7 | [34] |
| OMC–GNPs/L-Lys/GCE | 200 pM | 5 × 10−10–5 × 10−5 | [35] |
| DNAzyme/depAu/GCE | 33 fM | 10-13–2 × 10−7 | [36] |
| TSP/Au | 80 pM | 10−11–10−6 | [37] |
| primer-AuNPs-S3/Pb2+ /MCH/S2/S1/depAu/GCE | 3.3 fM | 10−15–10−11 | [13] |
| Apt/ERGO/GCE | 0.51 fM | 10−15–10−9 | This work |
| Sample | Added (M) | Found (M) | RSD (%) | Apparent Recovery Factors (%) |
|---|---|---|---|---|
| Tap water | - | Not detected | - | - |
| 1.00 × 10−15 | 0.98 × 10−15 | 2.04 | 98 | |
| 1.00 × 10−12 | 1.10 × 10−12 | 2.57 | 110 | |
| 1.00 × 10−9 | 1.03 × 10−9 | 2.41 | 103 | |
| Valley water | - | Not detected | - | - |
| 1.00 × 10−15 | 0.97 × 10−15 | 1.82 | 97 | |
| 1.00 × 10−12 | 0.95 × 10−12 | 2.55 | 95 | |
| 1.00 × 10−9 | 1.03 × 10−9 | 1.85 | 103 | |
| Secondary treated wastewater a | - | Not detected | - | - |
| 1.00 × 10−15 | 1.05 × 10−15 | 1.99 | 105 | |
| 1.00 × 10−12 | 1.03 × 10−12 | 1.27 | 103 | |
| 1.00 × 10−9 | 1.05 × 10−9 | 1.85 | 105 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, S.H.; Lee, C.-S.; Kim, T.H. Electrochemical Detection of Ultratrace Lead Ion through Attaching and Detaching DNA Aptamer from Electrochemically Reduced Graphene Oxide Electrode. Nanomaterials 2019, 9, 817. https://0-doi-org.brum.beds.ac.uk/10.3390/nano9060817
Yu SH, Lee C-S, Kim TH. Electrochemical Detection of Ultratrace Lead Ion through Attaching and Detaching DNA Aptamer from Electrochemically Reduced Graphene Oxide Electrode. Nanomaterials. 2019; 9(6):817. https://0-doi-org.brum.beds.ac.uk/10.3390/nano9060817
Chicago/Turabian StyleYu, Su Hwan, Chang-Seuk Lee, and Tae Hyun Kim. 2019. "Electrochemical Detection of Ultratrace Lead Ion through Attaching and Detaching DNA Aptamer from Electrochemically Reduced Graphene Oxide Electrode" Nanomaterials 9, no. 6: 817. https://0-doi-org.brum.beds.ac.uk/10.3390/nano9060817