
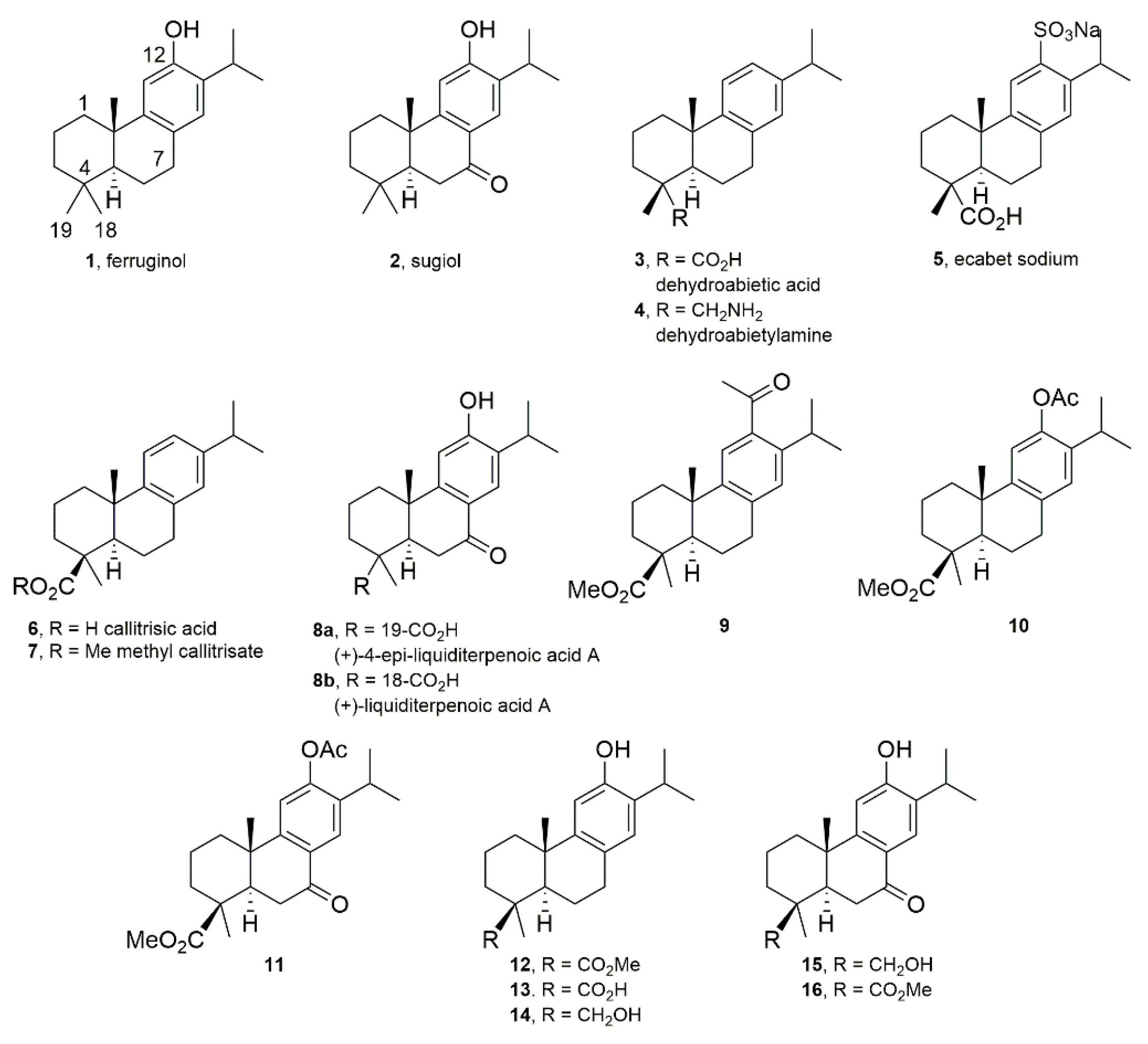
Biological Profiling of Semisynthetic C19-Functionalized Ferruginol and Sugiol Analogues

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
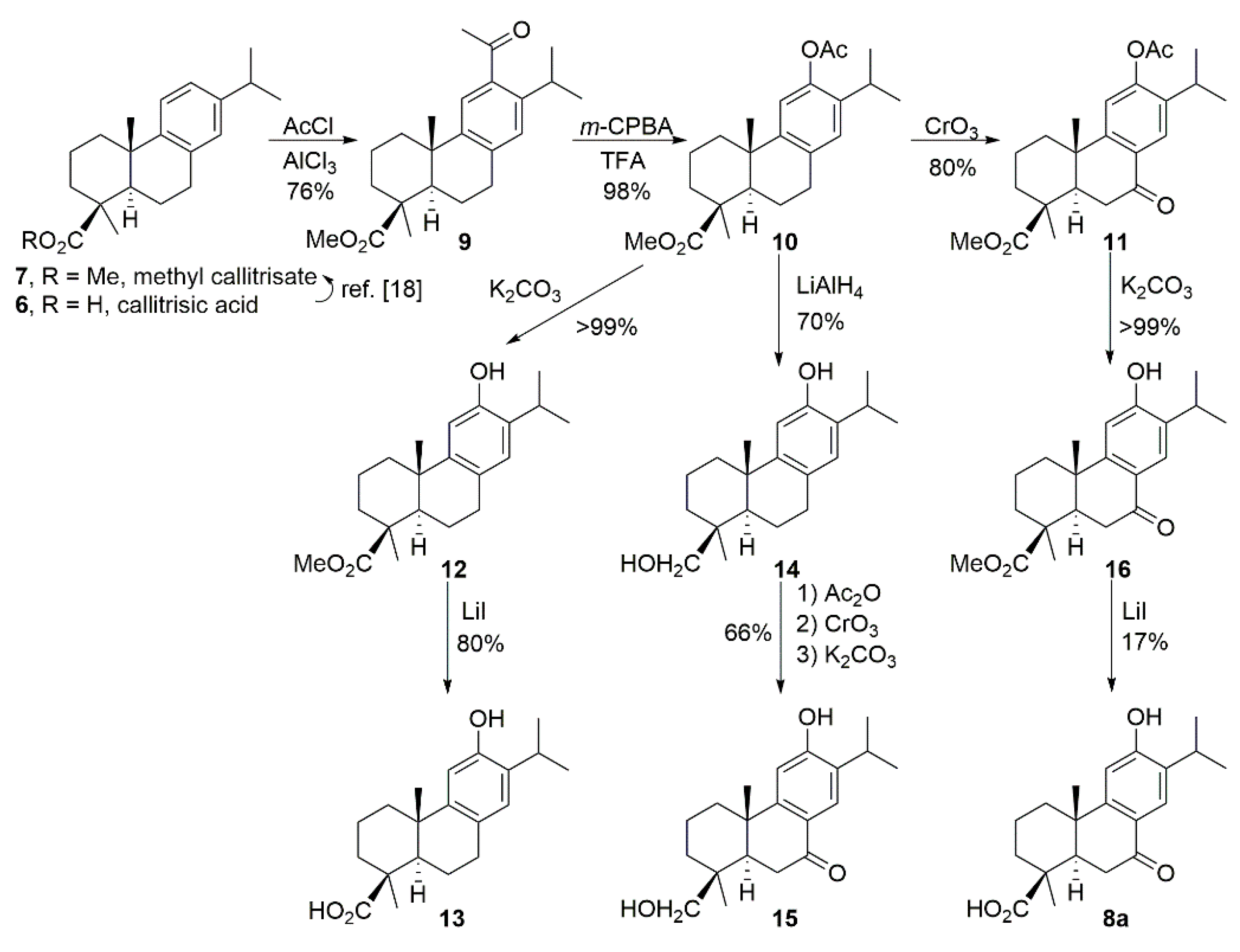
2.1. Chemistry
2.2. Biology
2.2.1. Antiparasitic Activity
2.2.2. Antiproliferative Activity
2.2.3. GABAA Receptor Modulating Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Chemistry
3.3. Biology
3.3.1. Antileishmanial Activity
3.3.2. Antiproliferative Activity
3.3.3. Expression of GABAA Receptors in Xenopus Laevis Oocytes
3.3.4. Antimalarial Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Singh, N.; Mishra, B.B.; Bajpai, S.; Singh, R.K.; Tiwari, V.K. Natural product based leads to fight leishmaniasis. Bioorg. Med. Chem. 2014, 22, 18–45. [Google Scholar] [CrossRef]
- González, M.A. Synthetic derivatives of aromatic abietane diterpenoids and their biological activities. Eur. J. Med. Chem. 2014, 87, 834–842. [Google Scholar] [CrossRef]
- González, M.A. Aromatic abietane diterpenoids: Their biological activity and synthesis. Nat. Prod. Rep. 2015, 32, 684–704. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.-T.; Tung, Y.-T.; Kuo, Y.-H.; Lin, C.-C.; Wu, J.-H. Ferruginol Inhibits Non–Small Cell Lung Cancer Growth by Inducing Caspase-Associated Apoptosis. Integr. Cancer Ther. 2015, 14, 86–97. [Google Scholar] [CrossRef]
- Xiong, W.-D.; Gong, J.; Xing, C. Ferruginol exhibits anticancer effects in OVCAR-3 human ovary cancer cells by inducing apoptosis, inhibition of cancer cell migration and G2/M phase cell cycle arrest. Mol. Med. Rep. 2017, 16, 7013–7017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Wu, C.; Zhang, B.; Zhang, Y.; Li, J. Ferruginol induced apoptosis on SK-Mel-28 human malignant melanoma cells mediated through P-p38 and NF-κB. Hum. Exp. Toxicol. 2019, 38, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Zhou, J.; Li, G.; Hu, N.; Xia, X.; Zhou, H. Ferruginol Diterpenoid Selectively Inhibits Human Thyroid Cancer Growth by Inducing Mitochondrial Dependent Apoptosis, Endogenous Reactive Oxygen Species (ROS) Production, Mitochondrial Membrane Potential Loss and Suppression of Mitogen-Activated Protein Kinase (MAPK) and PI3K/AKT Signaling Pathways. Med. Sci. Monit. 2019, 25, 2935–2942. [Google Scholar] [CrossRef]
- Fronza, M.; Murillo, R.; Slusarczyk, S.; Adams, M.; Hamburguer, M.; Heinzmann, B.; Laufer, S.; Merfort, I. In vitro cytotoxic activity of abietane diterpenes from Peltodon longipes as well as Salvia miltiorrhiza and Salvia sahendica. Bioorg. Med. Chem. 2011, 19, 4876–4881. [Google Scholar] [CrossRef]
- Iwamoto, M.; Minami, T.; Tokuda, H.; Ohtsu, H.; Tanaka, R. Potential Antitumor Promoting Diterpenoids from the Stem Bark of Thuja standishii. Planta Med. 2003, 69, 69–72. [Google Scholar] [CrossRef]
- Jung, S.-N.; Shin, D.-S.; Kim, H.-N.; Jeon, Y.J.; Yun, J.; Lee, Y.-J.; Kang, J.S.; Han, D.C.; Kwon, B.-M. Sugiol inhibits STAT3 activity via regulation of transketolase and ROS-mediated ERK activation in DU145 prostate carcinoma cells. Biochem. Pharmacol. 2015, 97, 38–50. [Google Scholar] [CrossRef]
- Gonçalves, M.D.; Bortoleti, B.T.S.; Tomiotto-Pellissier, F.; Miranda-Sapla, M.M.; Assolini, J.P.; Carloto, A.C.M.; Carvalho, P.G.C.; Tudisco, E.T.; Urbano, A.; Ambrósio, S.R.; et al. Dehydroabietic acid isolated from Pinus elliottii exerts in vitro antileishmanial action by pro-oxidant effect, inducing ROS production in promastigote and downregulating Nrf2/ferritin expression in amastigote forms of Leishmania amazonensis. Fitoterapia 2018, 128, 224–232. [Google Scholar] [CrossRef]
- Vahermo, M.; Krogerus, S.; Nasereddin, A.; Kaiser, M.; Brun, R.; Jaffe, C.L.; Yli-Kauhaluoma, J.; Moreira, V.M. Antiprotozoal activity of dehydroabietic acid derivatives against Leishmania donovani and Trypanosoma cruzi. Med. Chem. Commun. 2016, 7, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Pirttimaa, M.; Nasereddin, A.; Kopelyanskiy, D.; Kaiser, M.; Yli-Kauhaluoma, J.; Oksman-Caldentey, K.-M.; Brun, R.; Jaffe, C.L.; Moreira, V.M.; Alakurtti, S. Abietane-type diterpenoid amides with highly potent and selective activity against Leishmania donovani and Trypanosoma cruzi. J. Nat. Prod. 2016, 79, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Samoylenko, V.; Dunbar, D.C.; Gafur, M.A.; Khan, S.I.; Ross, S.A.; Mossa, J.S.; El-Feraly, F.S.; Tekwani, B.L.; Bosselaers, J.; Muhammad, I. Antiparasitic, nematicidal and antifouling constituents from Juniperus berries. Phytother. Res. 2008, 22, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Rueda, D.C.; Raith, M.; De Mieri, M.; Schöffmann, A.; Hering, S.; Hamburger, M. Identification of dehydroabietc acid from Boswellia thurifera resin as a positive GABAA receptor modulator. Fitoterapia 2014, 99, 28–34. [Google Scholar] [CrossRef]
- Hamulić, D.; Stadler, M.; Hering, S.; Padrón, J.M.; Bassett, R.; Rivas, F.; Loza-Mejía, M.A.; Dea-Ayuela, M.A.; González-Cardenete, M.A. Synthesis and Biological Studies of (+)-Liquiditerpenoic Acid A (Abietopinoic Acid) and Representative Analogues: SAR Studies. J. Nat. Prod. 2019, 82, 823–831. [Google Scholar] [CrossRef] [Green Version]
- González, M.A.; Zaragozá, R.J. Semisynthesis of the antiviral abietane diterpenoid jiadifenoic acid C from callitrisic acid (4-epidehydroabietic acid) isolated from sandarac resin. J. Nat. Prod. 2014, 77, 2114–2117. [Google Scholar] [CrossRef]
- González-Cardenete, M.A.; Zaragozá, R.J. A Short and Improved Synthesis of the Antiprotozoal Abietane Diterpenoid (−)-Sugikurojin A. Synthesis 2017, 49, 1315–1318. [Google Scholar] [CrossRef]
- Dea-Ayuela, M.A.; Bilbao-Ramos, P.; Bolás-Fernández, F.; González-Cardenete, M.A. Synthesis and antileishmanial activity of C7- and C12-functionalized dehydroabietylamine derivatives. Eur. J. Med. Chem. 2016, 121, 445–450. [Google Scholar] [CrossRef]
- Guiguemde, W.A.; Shelat, A.A.; Bouck, D.; Duffy, S.; Crowther, G.J.; Davis, P.H.; Smithson, D.C.; Connelly, M.; Clark, J.; Zhu, F.; et al. Chemical genetics of Plasmodium falciparum. Nature 2010, 465, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Nwaka, S.; Hudson, A. Innovative lead discovery strategies for tropical diseases. Nat. Rev. Drug Discov. 2006, 5, 941–955. [Google Scholar] [CrossRef]
- González, M.A.; Clark, J.; Connelly, M.; Rivas, F. Antimalarial activity of abietane ferruginol analogues possessing a phthalimide group. Bioorg. Med. Chem. Lett. 2014, 24, 5234–5237. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Li, S.; Wang, P.; Deng, G.; Yuan, W.; Su, Z. Cytotoxic compounds from invasive giant salvinia (Salvinia molesta) against human tumor cells. Bioorg. Med. Chem. Lett. 2013, 23, 6682–6687. [Google Scholar] [CrossRef]
- Stadler, M.; Padrón, J.M.; González-Cardenete, M.A. Antiproliferative Activity and Effect on GABAA Receptors of Callitrisic Acid Derivatives. Planta Med. Int. Open 2017, 4, e89–e92. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-C.; Wu, X.-D.; He, J.; Li, Y.; Zhang, R.-P.; Zhao, Q.-S. Three new abietane diterpenoids from Podocarpus fleuryi. Phytochem. Lett. 2013, 6, 364–367. [Google Scholar] [CrossRef]
- Bilbao-Ramos, P.; Galiana-Roselló, C.; Dea-Ayuela, M.A.; González-Alvarez, M.; Vega, C.; Rolón, M.; Pérez-Serrano, J.; Bolás-Fernández, F.; González-Rosende, M.E. Nuclease activity and ultrastructural effects of new sulfonamides with anti-leishmanial and trypanocidal activities. Parasitol. Int. 2012, 61, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Bilbao-Ramos, P.; Sifontes-Rodríguez, S.; Dea-Ayuela, M.A.; Bolás-Fernández, F. A fluorometric method for evaluation of pharmacological activity against intracellular Leishmania amastigotes. J. Microbiol. Methods 2012, 89, 8–11. [Google Scholar] [CrossRef]
- Galiana-Roselló, C.; Bilbao-Ramos, P.; Dea-Ayuela, M.A.; Rolón, M.; Vega, C.; Bolás-Fernández, F.; García-España, E.; Alfonso, J.; Coronel, C.; González-Rosende, M.E. In vitro and in vivo antileishmanial and trypanocidal studies of new N-benzene- and N-naphthalenesulfonamide derivatives. J. Med. Chem. 2013, 56, 8984–8998. [Google Scholar] [CrossRef]
- Luger, D.; Poli, G.; Wieder, M.; Stadler, M.; Ke, S.; Ernst, M.; Hohaus, A.; Linder, T.; Seidel, T.; Langer, T.; et al. Identification of the putative binding pocket of valerenic acid on GABAA receptors using docking studies and site-directed mutagenesis. Br. J. Pharmacol. 2015, 172, 5403–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tipparaju, S.K.; Joyasawal, S.; Pieroni, M.; Kaiser, M.; Brun, R.; Kozikowski, A.P. In Pursuit of Natural Product Leads: Synthesis and Biological Evaluation of 2-[3-hydroxy-2-[(3-hydroxypyridine-2-carbonyl)amino]phenyl]benzoxazole-4-carboxylic acid (A-33853) and Its Analogues: Discovery of N-(2-Benzoxazol-2-ylphenyl)benzamides as Novel Antileishmanial Chemotypes. J. Med. Chem. 2008, 51, 7344–7347. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compound | L. infantum | L. donovani | L. amazonensis | L. guyanensis | Macrophages J774 | ||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 a ± SD | SI b | IC50 ± SD | SI | IC50 ± SD | SI | IC50 ± SD | SI | CC50 | |
| 1d | - | - | 12.2 | >1.4 | - | - | - | - | >16.6 (Vero) |
| 8a | 100.3 ± 20.0 | >2.0 | 84.4 ± 31.6 | >2.4 | 94.5 ± 6.5 | >2.1 | 3.0 ± 0.9 | >67.0 | >200 |
| 8be | NA f | NA f | NA f | NA f | 219.4 ± 15.0 | ||||
| 9 | 20.3 ± 4.6 | >10.0 | 8.9 ± 2.0 | >22.0 | 14.1 ± 1.4 | >14.0 | 18.7 ± 0.8 | >10.7 | >200 |
| 9be | 2.5 ± 0.6 | 51.8 | 14.8 ± 0.9 | 8.8 | 11.6 ± 0.6 | 11.2 | 14.2 ± 0.4 | 9.1 | 129.6 ± 8.9 |
| 10 | 3.6 ± 0.6 | >8.4 | 3.9 ± 0.1 | >7.7 | 7.4 ± 0.6 | >4.1 | ND g | - | 30.2 ± 2.0 |
| 10be | 1.3 ± 0.1 | 20.4 | 8.1 ± 0.7 | 3.3 | 3.2 ± 0.3 | 8.2 | 5.5 ± 0.7 | 4.8 | 26.5 ± 2.7 |
| 11 | ND g | - | ND g | - | ND g | - | ND g | - | 45.8 ± 5.6 |
| 11be | 0.7 ± 0.2 | 25.4 | 4.4 ± 0.5 | 4.1 | 0.65 ± 0.03 | 27.4 | 1.3 ± 0.1 | 13.6 | 17.8 ± 2.8 |
| 12 | ND g | - | ND g | - | ND g | - | ND g | - | 65.4 ± 7.0 |
| 12be | 5.0 ± 0.3 | 6.8 | 9.21 ± 0.06 | 3.7 | 3.9 ± 0.1 | 8.7 | 5.9 ± 1.1 | 5.8 | 34.2 ± 3.1 |
| 13 | 29.1 ± 1.5 | 7.5 | 12.8 ± 2.8 | 17.0 | 51.5 ± 3.2 | 4.2 | 17.8 ± 1.2 | 12.2 | 217.4 ± 34.0 |
| 13be | 43.6 ± 3.4 | 2.0 | 58.1 ± 0.7 | 1.5 | 54.1 ± 6.5 | 1.6 | 71.4 ± 1.5 | 1.2 | 85.8 ± 28.8 |
| 14 | ND g | - | ND g | - | ND g | - | ND g | - | 51.9 ± 5.9 |
| 14be | 0.7 ± 0.1 | 33.4 | 13.9 ± 0.1 | 1.7 | 7.7 ± 0.8 | 3.0 | 12.5 ± 2.2 | 1.9 | 23.4 ± 4.6 |
| 15 | ND g | - | ND g | - | ND g | - | ND g | - | 50.1 ± 2.4 |
| 15be | 9.7 ± 0.8 | 1.5 | 19.7 ± 0.4 | 0.7 | 10.2 ± 0.6 | 1.4 | 16.5 ± 0.7 | 0.9 | 14.4 ± 2.8 |
| 16 | 10.1 ± 1.4 | >19.8 | 11.8 ± 0.8 | >16.9 | 8.5 ± 0.3 | >23.5 | 5.6 ± 0.6 | >35.7 | >200 |
| 16be | ND g | ND g | ND g | ND g | 153.9 ± 49.3 | ||||
| Miltefosine | 3.4 ± 0.6 | 40.1 | 0.15 ± 0.02 | 909 | 47.7 ± 5.0 | 2.9 | 18.2 ± 0.6 | 7.5 | 136.4 ± 1.4 |
| Compound | L. infantum | L. amazonensis | Macrophages J774 | ||
|---|---|---|---|---|---|
| IC50 a ± SD | SI b | IC50 ± SD | SI | CC50 c | |
| 8a | 20.1 ± 2.0 | >9.9 | 21.4 ± 2.3 | >9.3 | >200 |
| 9 | 22.7 ± 5.9 | >8.8 | 30.3 ± 1.2 | >6.6 | >200 |
| 9bd | 37.2 ± 2.4 | 3.5 | 31.4 ± 6.2 | 4.1 | 129.6 ± 8.9 |
| 13 | 19.2 ± 3.8 | >11.3 | 8.8 ± 1.6 | >24.7 | 217.4 ± 34.0 |
| 16 | 16.5 ± 4.6 | >12.1 | 16.1 ± 2.0 | >12.4 | >200 |
| Miltefosine | 58.1 ± 4.4 | 2.3 | 16.5 ± 1.9 | 8.2 | 136.4 ± 1.4 |
| Compound | P. falciparuma | Cell Lines b | ||||||
|---|---|---|---|---|---|---|---|---|
| 3D7 | K1 | HepG2 | RAJI | BJ | HEK293 | |||
| EC50 c | SI d | EC50 c | SI d | EC50 c | EC50 c | EC50 c | EC50 c | |
| 1e | 2.47 | 4.6 | 1.33 | 8.6 | >25 | 11.49 | 19.57 | >25 |
| 8a | >15 | n.c. | >15 | n.c. | >25 | >25 | >25 | >25 |
| 9 | 12.8 | 1.9 | 12.6 | 2.0 | >25 | >25 | >25 | >25 |
| 10 | >15 | n.c. | 3.1 | 8.1 | >25 | >25 | >25 | >25 |
| 11 | 7.1 | 3.5 | 5.7 | 4.4 | >25 | >25 | >25 | >25 |
| 12 | 4.7 | 5.3 | 2.1 | 11.9 | >25 | >25 | >25 | >25 |
| 13 | >15 | n.c. | >15 | n.c. | >25 | >25 | >25 | >25 |
| 14 | >15 | n.c. | 11.6 | 2.2 | >25 | >25 | >25 | >25 |
| 15 | >15 | n.c. | 9.4 | 2.6 | >25 | >25 | >25 | >25 |
| 16 | 4.8 | 5.2 | 3.9 | 6.4 | >25 | >25 | >25 | >25 |
| Chloroquine | 0.023 | n.c. | 0.69 | n.c. | >25 | >25 | >25 | >25 |
| Gambogic acid | - | - | - | - | >25 | 0.24 | 0.21 | 0.21 |
| Staurosporine | - | - | - | - | <0.2 | <0.2 | <0.2 | <0.2 |
| Compound | Cell Line (Origin) | |||||
|---|---|---|---|---|---|---|
| A549 (Lung) | HBL-100 (Breast) | HeLa (Cervix) | SW1573 (Lung) | T-47D (Breast) | WiDr (Colon) | |
| 1b | 30.7 ± 5.2 | - | - | - | - | - |
| 2b | 79.8 ± 2.2 | - | - | - | - | - |
| 7c | 10.0 ± 1.9 | 14.0 ± 5.0 | 16.0 ± 2.5 | 17.0 ± 2.2 | 8.8 ± 2.7 | 6.4 ± 2.1 |
| 8a | 12.0 ± 2.9 | 39.0 ± 14.0 | 12.0 ± 3.8 | 41.0 ± 17.0 | 31.0 ± 12.0 | 33.0 ± 15.0 |
| 8bd | >100 | >100 | 85.0 ± 21.0 | >100 | >100 | >100 |
| 9 | 15.0 ± 7.0 | 14.0 ± 2.1 | 15.0 ± 4.1 | 25.0 ± 11.0 | 14.0 ± 1.0 | 15.0 ± 2.1 |
| 9bd | 18.0 ± 2.3 | 15.0 ± 1.2 | 11.0 ± 1.0 | 11.0 ± 4.6 | 10.0 ± 1.5 | 17.0 ± 2.4 |
| 10 | 6.5 ± 2.7 | 11.0 ± 2.3 | 7.0 ± 1.7 | 18.0 ± 3.4 | 4.2 ± 1.5 | 4.5 ± 1.4 |
| 10bd | 11.0 ± 1.8 | 15.0 ± 3.9 | 4.4 ± 1.1 | 12.0 ± 3.2 | 14.0 ± 2.9 | 16.0 ± 2.5 |
| 11 | 3.1 ± 0.9 | 7.4 ± 0.7 | 5.6 ± 2.9 | 17.0 ± 7.5 | 6.1 ± 1.9 | 5.2 ± 3.1 |
| 11bd | 14.0 ± 0.4 | 16.0 ± 1.5 | 13.0 ± 2.3 | 17.0 ± 1.9 | 20.0 ± 2.9 | 19.0 ± 1.7 |
| 12 | 2.3 ± 1.5 | 11.0 ± 3.0 | 5.1 ± 1.9 | 12.0 ± 4.1 | 4.2 ± 1.3 | 3.7 ± 1.9 |
| 12bd | 9.4 ± 2.6 | 9.7 ± 2.7 | 5.8 ± 0.5 | 11.0 ± 2.0 | 19.0 ± 3.8 | 23.0 ± 4.4 |
| 13 | 17.0 ± 6.1 | 34.0 ± 4.2 | 29.0 ± 13.0 | 32.0 ± 8.9 | 41.0 ± 8.4 | 59.0 ± 7.3 |
| 13bd | 70.0 ± 6.3 | >100 | 96.0 ± 5.5 | 93.0 ± 9.3 | >100 | >100 |
| 14 | 1.8 ± 0.4 | 13.0 ± 1.6 | 3.1 ± 0.9 | 13.0 ± 1.3 | 12.0 ± 1.8 | 13.0 ± 3.7 |
| 14bd | 12.0 ± 1.9 | 16.0 ± 2.0 | 12.0 ± 1.8 | 15.0 ± 2.8 | 16.0 ± 0.9 | 17.0 ± 1.3 |
| 15 | 10.0 ± 0.3 | 17.0 ± 1.7 | 15.0 ± 1.1 | 19.0 ± 3.3 | 19.0 ± 1.1 | 17.0 ± 5.0 |
| 15bd | 19.0 ± 3.3 | 16.0 ± 2.0 | 16.0 ± 2.4 | 23.0 ± 0.7 | 20.0 ± 2.7 | 19.0 ± 2.1 |
| 16 | 11.0 ± 6.9 | 65.0 ± 4.9 | 18.0 ± 1.7 | >100.0 | 47.0 ± 25.0 | 6.1 ± 4.2 |
| 16bd | >100 | >100 | >100 | >100 | >100 | >100 |
| etoposide | 1.5 ± 0.3 | 1.4 ± 0.1 | 3.3 ± 1.6 | 15.0 ± 1.5 | 22.0 ± 5.5 | 23.0 ± 3.1 |
| cisplatin | 4.9 ± 0.2 | 1.9 ± 0.2 | 1.8 ± 0.5 | 2.7 ± 0.4 | 17.0 ± 3.3 | 23.0 ± 4.3 |
| Compound | IGABA Potentiation of 10 µM (%) | IGABA Potentiation of 100 µM (%) |
|---|---|---|
| Dehydroabietic acid (3) | 192 ± 10 | 789 ± 82 |
| Callitrisic acid (6) a | 14 ± 5 | 269 ± 24 |
| 8a | −8 ± 1 | −8 ± 3 |
| 8bb | −2.2 ± 4.4 | −21.1 ± 7.1 |
| 9 | - | - |
| 9bb | 2.7 ± 0.9 | −2.9 ± 5.1 |
| 10 | - | - |
| 10bb | 5.1 ± 2.1 | 26.3 ± 8.9 |
| 11 | 1 ± 3 | 75 ± 4 |
| 11bb | −1.0 ± 2.8 | 46.7 ± 8.3 |
| 12 | 1 ± 3 | 65 ± 6 |
| 12bb | 23.7 ± 9.6 | 378.0 ± 65.6 |
| 13 | 3 ± 3 | 71 ± 23 |
| 13bb | 8.9 ± 2.3 | 12.1 ± 2.9 |
| 14 | 64 ± 13 | 507 ± 102 |
| 14bb | 29.3 ± 10.6 | 154.1 ± 34.2 |
| 15 | 30 ± 14 | 238 ± 28 |
| 15bb | 4.5 ± 5.8 | −30.3 ± 9.5 |
| 16 | 5 ± 4 | 104 ± 26 |
| 16bb | 37.4 ± 12.7 | 242.4 ± 46.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Cardenete, M.A.; Rivas, F.; Basset, R.; Stadler, M.; Hering, S.; Padrón, J.M.; Zaragozá, R.J.; Dea-Ayuela, M.A. Biological Profiling of Semisynthetic C19-Functionalized Ferruginol and Sugiol Analogues. Antibiotics 2021, 10, 184. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020184
González-Cardenete MA, Rivas F, Basset R, Stadler M, Hering S, Padrón JM, Zaragozá RJ, Dea-Ayuela MA. Biological Profiling of Semisynthetic C19-Functionalized Ferruginol and Sugiol Analogues. Antibiotics. 2021; 10(2):184. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020184
Chicago/Turabian StyleGonzález-Cardenete, Miguel A., Fatima Rivas, Rachel Basset, Marco Stadler, Steffen Hering, José M. Padrón, Ramón J. Zaragozá, and María Auxiliadora Dea-Ayuela. 2021. "Biological Profiling of Semisynthetic C19-Functionalized Ferruginol and Sugiol Analogues" Antibiotics 10, no. 2: 184. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020184