
Weighted Gene Co-Expression Network Analysis Identifies Key Modules and Hub Genes Associated with Mycobacterial Infection of Human Macrophages




Abstract
:1. Introduction
2. Results
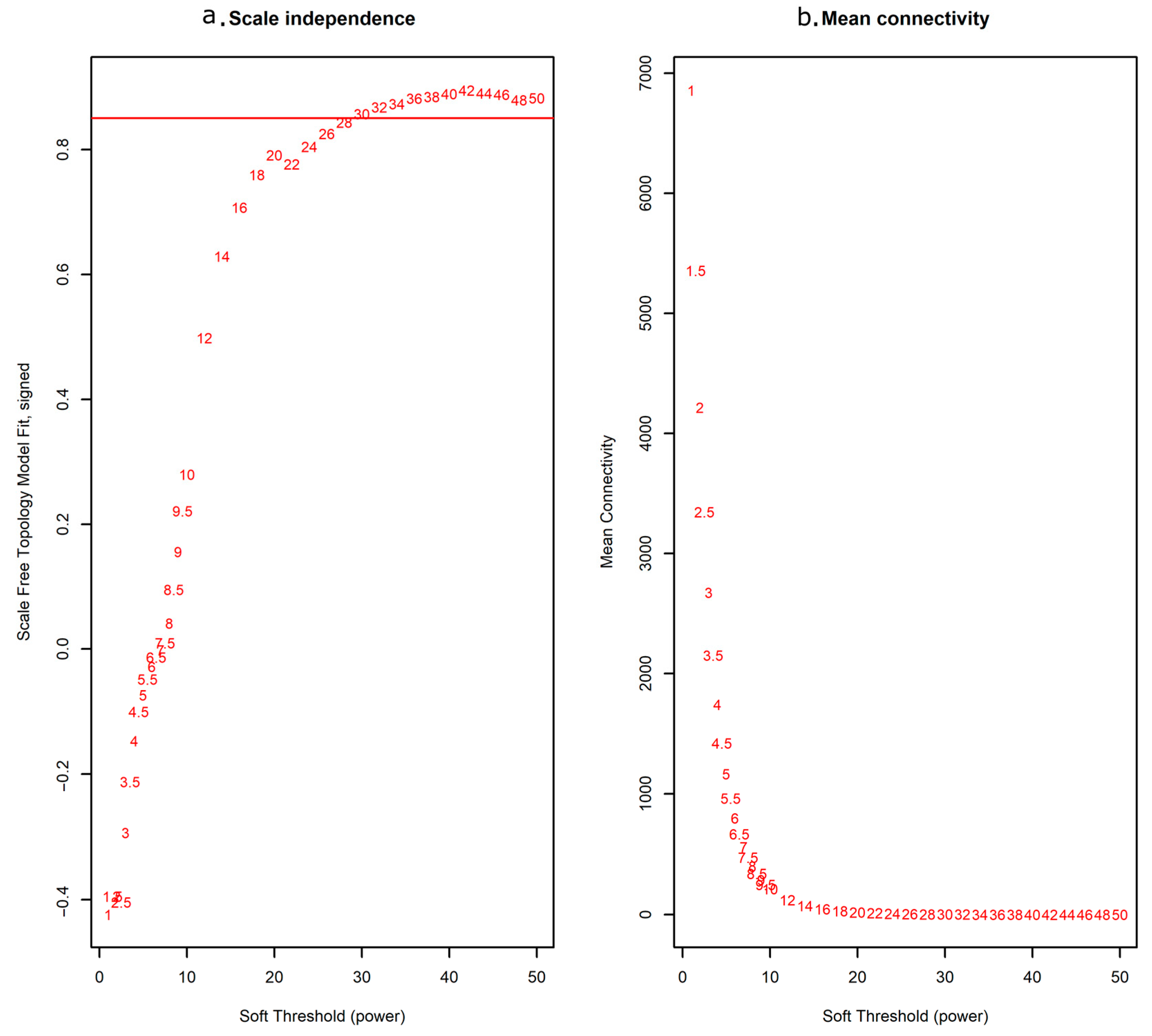
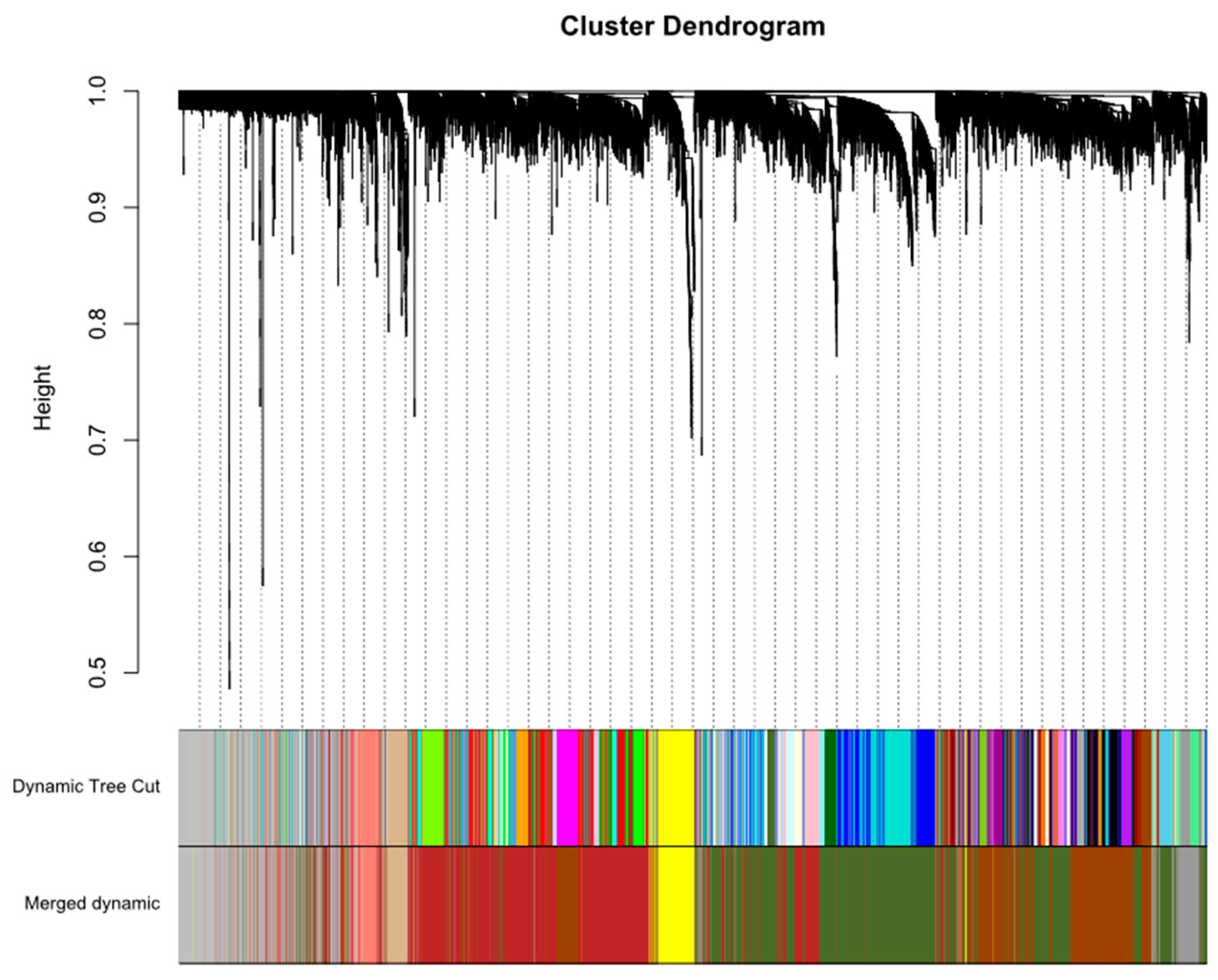
2.1. Construction of Weighted Co-Expression Network
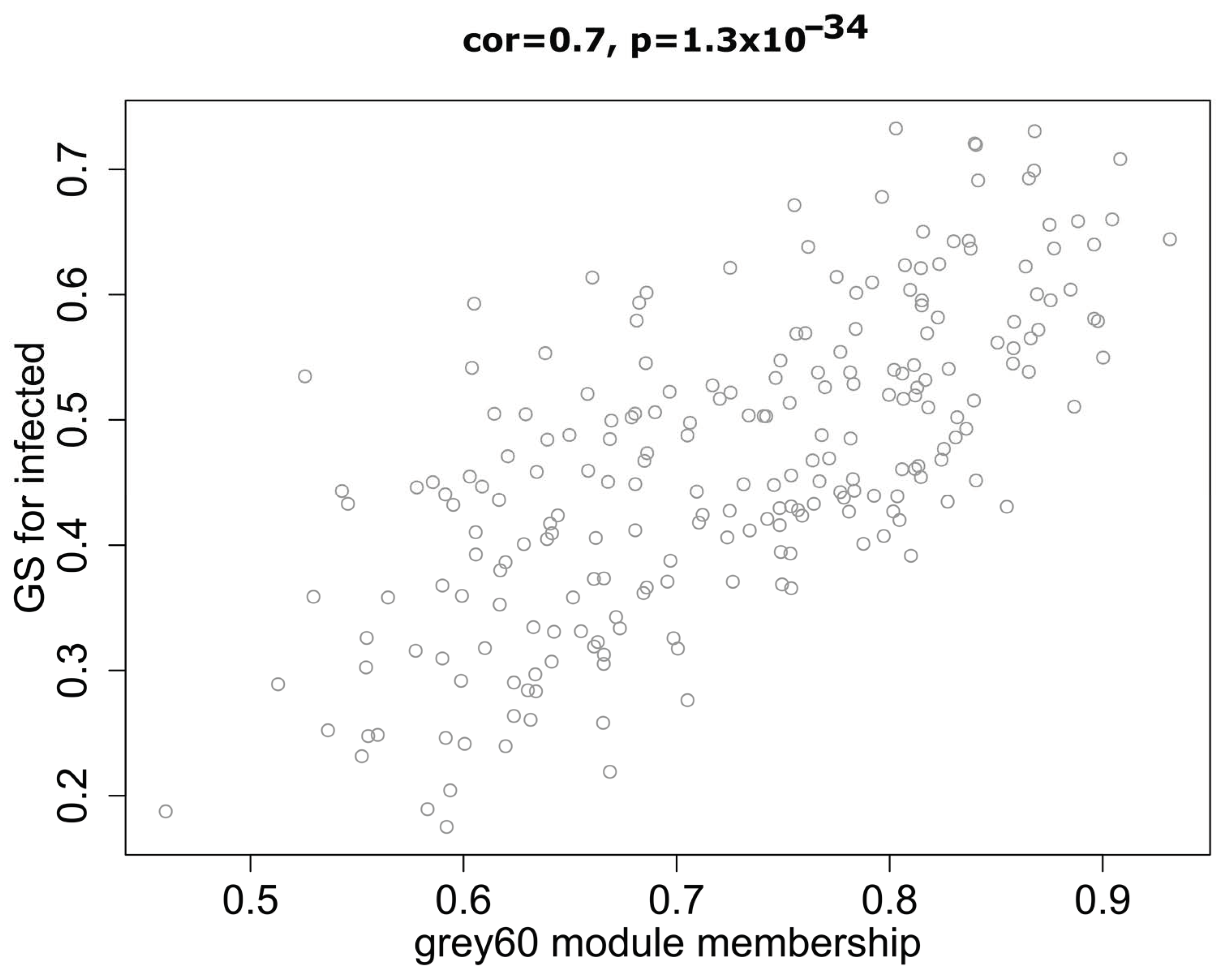
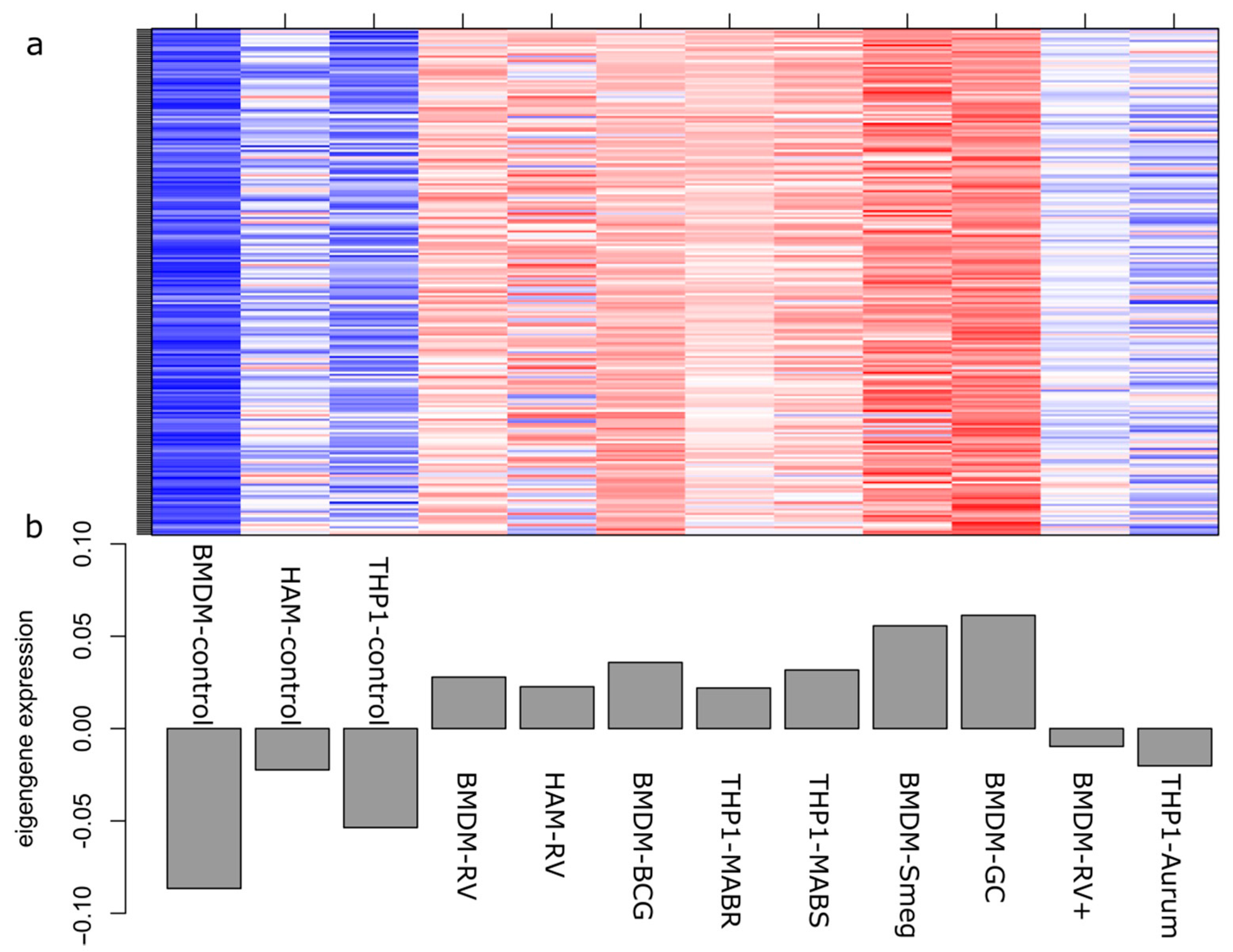
2.2. Correlation between Modules and Identification of Key Modules
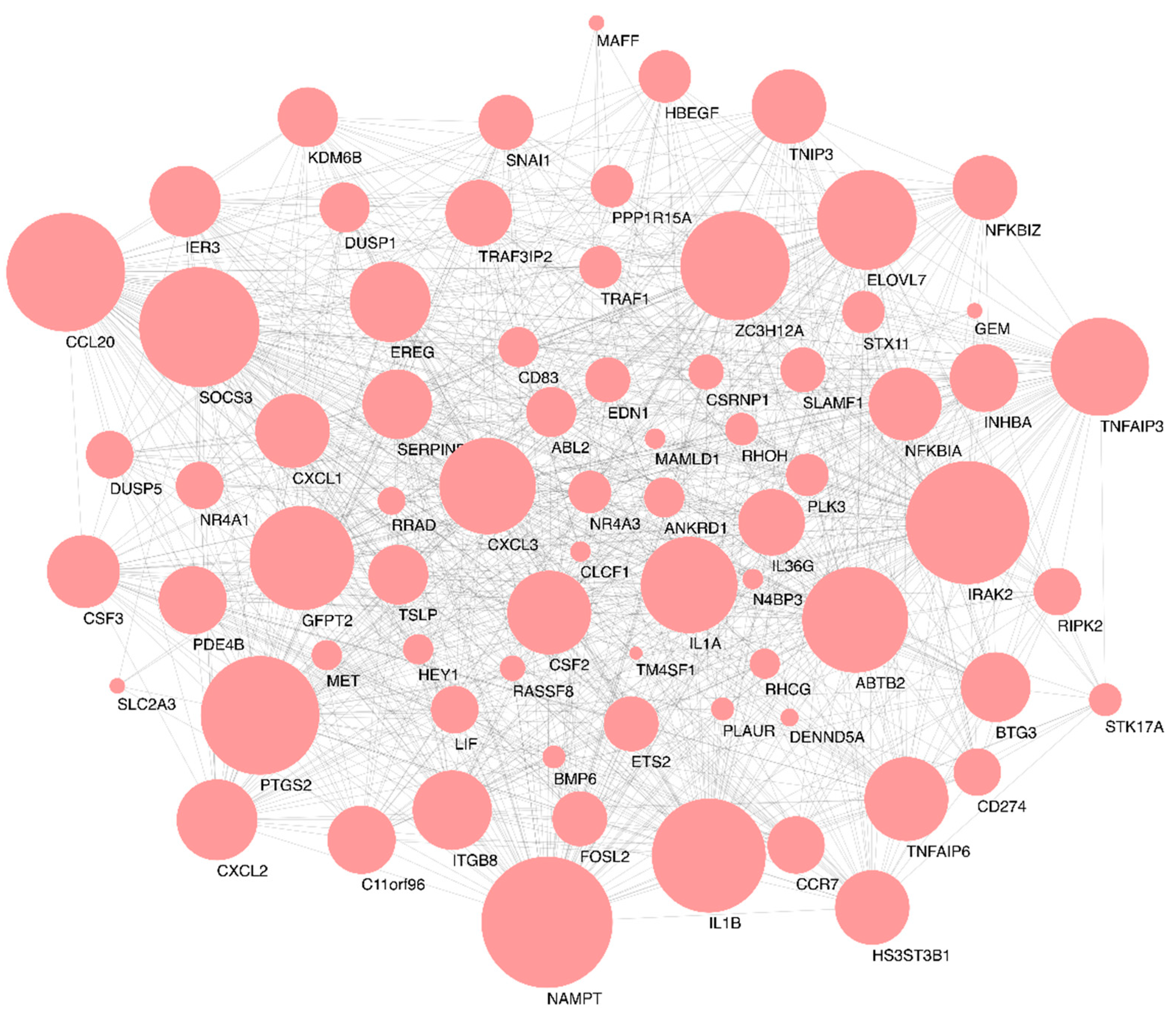
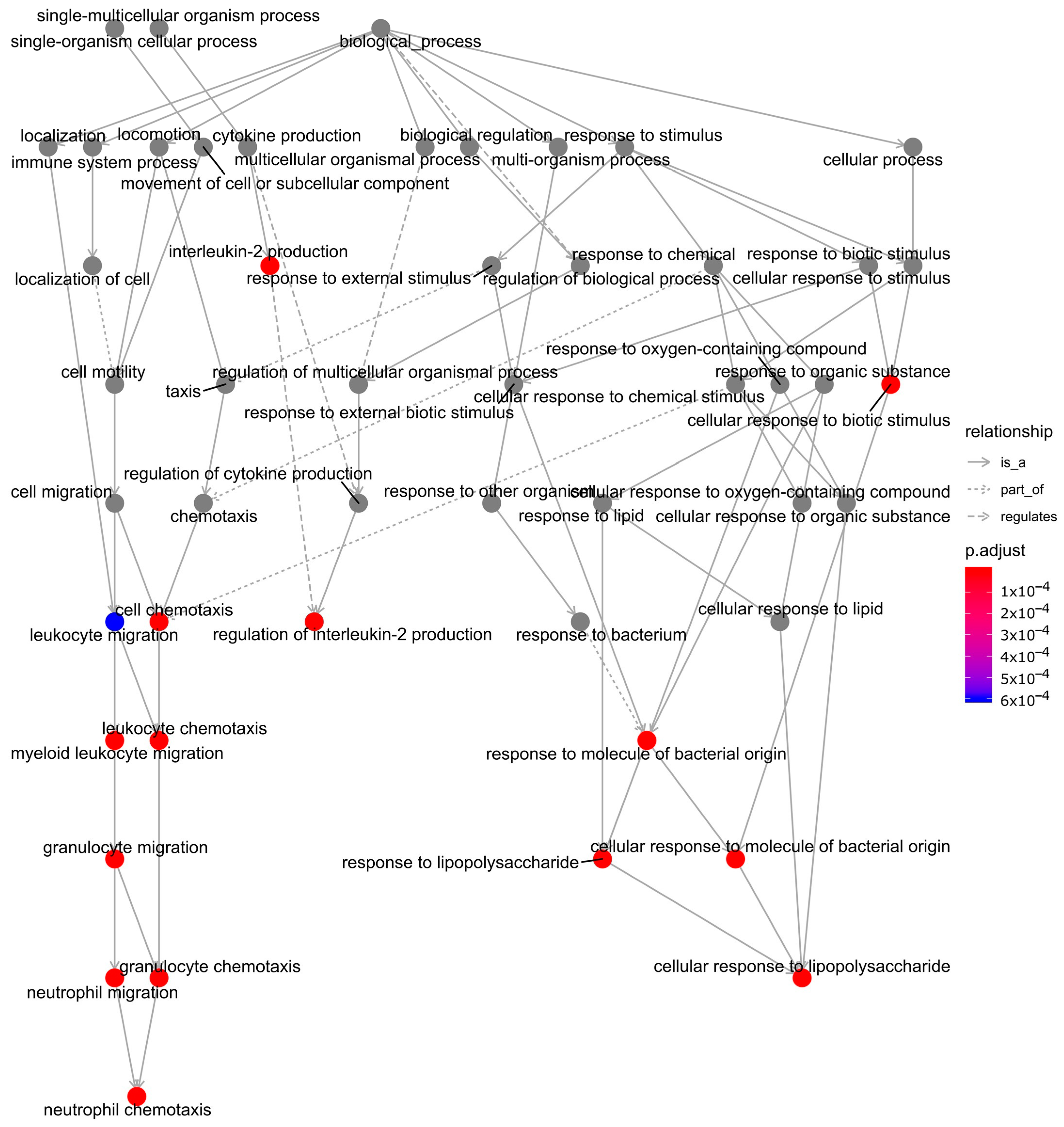
2.3. Functional Enrichment and Identification of Hub Genes
3. Discussion
4. Materials and Methods
4.1. RNA Sequencing of Human Monocyte-Derived Macrophage (THP-1) Infected with M. Aurum
4.2. Data Collection and Sample Processing for WGCNA
4.3. Construction of Weighted Gene Co-Expressed Networks
4.4. Functional Enrichment of Recurrence-Associated Modules
4.5. Identification of Hub Genes in Key Modules
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix B
Appendix C
Appendix D
Appendix E
References
- WHO. Global Tuberculosis Report 2019; WHO: Geneva, Switzerland, 2019; ISBN 9789241565714. [Google Scholar]
- Gupta, A.; Kaul, A.; Tsolaki, A.G.; Kishore, U.; Bhakta, S. Mycobacterium tuberculosis: Immune evasion, latency and reactivation. Immunobiology 2012, 217, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Ferluga, J.; Yasmin, H.; Al-Ahdal, M.N.; Bhakta, S.; Kishore, U. Natural and trained innate immunity against Mycobacterium tuberculosis. Immunobiology 2020, 225, 151951. [Google Scholar] [CrossRef] [PubMed]
- Tufariello, J.M.; Chan, J.; Flynn, J.L. Latent tuberculosis: Mechanisms of host and bacillus that contribute to persistent infection. Lancet Infect. Dis. 2003, 3, 578–590. [Google Scholar] [CrossRef]
- Dey, B.; Bishai, W.R. Crosstalk between Mycobacterium tuberculosis and the host cell. Semin. Immunol. 2014, 26, 486–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connell, D.W.; Berry, M.; Cooke, G.; Kon, O.M. Update on tuberculosis: TB in the early 21st century. Eur. Respir. Rev. 2011, 20, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Corbett, E.L.; Watt, C.J.; Walker, N.; Maher, D.; Williams, B.G.; Raviglione, M.C.; Dye, C. The growing burden of tuberculosis: Global trends and interactions with the HIV epidemic. Arch. Intern. Med. 2003, 163, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Lohrasbi, V.; Talebi, M.; Bialvaei, A.Z.; Fattorini, L.; Drancourt, M.; Heidary, M.; Darban-Sarokhalil, D. Trends in the discovery of new drugs for Mycobacterium tuberculosis therapy with a glance at resistance. Tuberculosis 2018, 109, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Li, J.; Moussaoui, M.; Boix, E. Immune Modulation by Human Secreted RNases at the Extracellular Space. Front. Immunol. 2018, 9, 1012. [Google Scholar] [CrossRef] [Green Version]
- Coscolla, M.; Gagneux, S. Does M. tuberculosis genomic diversity explain disease diversity? Drug Discov. Today Dis. Mech. 2010, 7, e43–e59. [Google Scholar] [CrossRef] [Green Version]
- Correa-Macedo, W.; Cambri, G.; Schurr, E. The Interplay of Human and Mycobacterium Tuberculosis Genomic Variability. Front. Genet. 2019, 10, 865. [Google Scholar] [CrossRef]
- Jhingan, G.D.; Kumari, S.; Jamwal, S.V.; Kalam, H.; Arora, D.; Jain, N.; Kumaar, L.K.; Samal, A.; Rao, K.V.S.; Kumar, D.; et al. Comparative Proteomic Analyses of Avirulent, Virulent, and Clinical Strains of Mycobacterium tuberculosis Identify Strain-specific Patterns. J. Biol. Chem. 2016, 291, 14257–14273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blischak, J.D.; Tailleux, L.; Mitrano, A.; Barreiro, L.B.; Gilad, Y. Mycobacterial infection induces a specific human innate immune response. Sci. Rep. 2015, 5, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalpas, N.C.; de Park, S.; Magee, D.A.; Taraktsoglou, M.; Browne, J.A.; Conlon, K.M.; Rue-Albrecht, K.; Killick, K.E.; Hokamp, K.; Lohan, A.J.; et al. Whole-transcriptome, high-throughput RNA sequence analysis of the bovine macrophage response to Mycobacterium bovis infection in vitro. BMC Genom. 2013, 14, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malone, K.M.; Rue-Albrecht, K.; Magee, D.A.; Conlon, K.; Schubert, O.T.; Nalpas, N.C.; Browne, J.A.; Smyth, A.; Gormley, E.; Aebersold, R.; et al. Comparative ’omics analyses differentiate Mycobacterium tuberculosis and Mycobacterium bovis and reveal distinct macrophage responses to infection with the human and bovine tubercle bacilli. Microb. Genom. 2018, 4, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aulicino, A.; Dinan, A.M.; Miranda-CasoLuengo, A.A.; Browne, J.A.; Rue-Albrecht, K.; MacHugh, D.E.; Loftus, B.J. High-throughput transcriptomics reveals common and strain-specific responses of human macrophages to infection with Mycobacterium abscessus Smooth and Rough variants. BMC Genom. 2015, 16, 1046. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bhakta, S. An integrated surrogate model for screening of drugs against mycobacterium tuberculosis. J. Antimicrob. Chemother. 2012, 67, 1380–1391. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Lardone, R.D.; Chan, A.A.; Lee, A.F.; Foshag, L.J.; Faries, M.B.; Sieling, P.A.; Lee, D.J. Mycobacterium bovis Bacillus Calmette–Guérin Alters melanoma microenvironment Favoring antitumor T cell responses and improving M2 Macrophage Function. Front. Immunol. 2017, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Papp, A.C.; Azad, A.K.; Pietrzak, M.; Williams, A.; Handelman, S.K.; Igo, R.P.; Stein, C.M.; Hartmann, K.; Schlesinger, L.S.; Sadee, W. AmpliSeq transcriptome analysis of human alveolar and monocyte-derived macrophages over time in response to Mycobacterium tuberculosis infection. PLoS ONE 2018, 13, e0198221. [Google Scholar] [CrossRef]
- Coya, J.M.; De Matteis, L.; Giraud-Gatineau, A.; Biton, A.; Serrano-Sevilla, I.; Danckaert, A.; Dillies, M.-A.; Gicquel, B.; De la Fuente, J.M.; Tailleux, L. Tri-mannose grafting of chitosan nanocarriers remodels the macrophage response to bacterial infection. J. Nanobiotechnology 2019, 17, 15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Feng, H.; Li, Z.; Li, D.; Liu, S.; Huang, H.; Li, M. Application of weighted gene co-expression network analysis to identify key modules and hub genes in oral squamous cell carcinoma tumorigenesis. Onco. Targets. Ther. 2018, 11, 6001–6021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Huang, S.; Gao, H.; Zhou, Y.; Lai, C.; Li, Z.; Xian, W.; Qian, X.; Li, Z.; Huang, Y.; et al. Initial Whole Genome Sequencing and Analysis of the Host Genetic Contribution to COVID-19 Severity and Susceptibility. Cell Discov. 2020. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinforma. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Leprae, M.; Phelan, J.; Maitra, A.; Mcnerney, R.; Nair, M.; Gupta, A.; Coll, F.; Pain, A.; Bhakta, S.; Clark, T.G. The draft genome of Mycobacterium aurum, a potential model organism for investigating drugs against Mycobacterium tuberculosis and Mycobacterium leprae. Int. J. Mycobacteriology 2015, 4, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Namouchi, A.; Cimino, M.; Favre-Rochex, S.; Charles, P.; Gicquel, B. Phenotypic and genomic comparison of Mycobacterium aurum and surrogate model species to Mycobacterium tuberculosis: Implications for drug discovery. BMC Genom. 2017, 18, 530. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.P.; Chan, J.; Salgame, P. Mycobacterium tuberculosis Induces Differential Cytokine Production from Dendritic Cells and Macrophages with Divergent Effects on Naive T Cell Polarization. J. Immunol. 2002, 168, 4636–4642. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.M.; Mayer-Barber, K.D.; Sher, A. Role of innate cytokines in mycobacterial infection. Mucosal Immunol. 2011, 4, 252–260. [Google Scholar] [CrossRef]
- Van Crevel, R.; Ottenhoff, T.H.M.; van der Meer, J.W.M. Innate Immunity to Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2002, 15, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Boro, M.; Singh, V.; Balaji, K.N. Mycobacterium tuberculosis-triggered Hippo pathway orchestrates CXCL1/2 expression to modulate host immune responses. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Dunlap, M.D.; Howard, N.; Das, S.; Scott, N.; Ahmed, M.; Prince, O.; Rangel-Moreno, J.; Rosa, B.A.; Martin, J.; Kaushal, D.; et al. A novel role for C–C motif chemokine receptor 2 during infection with hypervirulent Mycobacterium tuberculosis. Mucosal Immunol. 2018, 11, 1727–1742. [Google Scholar] [CrossRef]
- Volpe, E.; Cappelli, G.; Grassi, M.; Martino, A.; Serafino, A.; Colizzi, V.; Sanarico, N.; Mariani, F. Gene expression profiling of human macrophages at late time of infection with Mycobacterium tuberculosis. Immunology 2006, 118, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The TLR and IL-1 signalling network at a glance. J. Cell Sci. 2014, 127, 2383–2390. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Wang, J.; Gao, G.F.; Liu, C.H. Insights into battles between Mycobacterium tuberculosis and macrophages. Protein Cell 2014, 5, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishrat, R. Stage specific classification of DEGs via statistical profiling and network analysis reveals potential biomarker associated with various stages of TB. bioRxiv 2018, 414110. [Google Scholar] [CrossRef] [Green Version]
- Ariel, O.; Gendron, D.; Dudemaine, P.L.; Gévry, N.; Ibeagha-Awemu, E.M.; Bissonnette, N. Transcriptome Profiling of Bovine Macrophages Infected by Mycobacterium avium spp. paratuberculosis Depicts Foam Cell and Innate Immune Tolerance Phenotypes. Front. Immunol. 2020, 10, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Goletti, D.; Lindestam Arlehamn, C.S.; Scriba, T.J.; Anthony, R.; Cirillo, D.M.; Alonzi, T.; Denkinger, C.M.; Cobelens, F. Can we predict tuberculosis cure? What tools are available? Eur. Respir. J. 2018, 52, 1801089. [Google Scholar] [CrossRef]
- Walzl, G.; Ronacher, K.; Hanekom, W.; Scriba, T.J.; Zumla, A. Immunological biomarkers of tuberculosis. Nat. Rev. Immunol. 2011, 11, 343–354. [Google Scholar] [CrossRef]
- Vrieling, F.; Kostidis, S.; Spaink, H.P.; Haks, M.C.; Mayboroda, O.A.; Ottenhoff, T.H.M.M.; Joosten, S.A. Analyzing the impact of Mycobacterium tuberculosis infection on primary human macrophages by combined exploratory and targeted metabolomics. Sci. Rep. 2020, 10, 7085. [Google Scholar] [CrossRef]
- Wu, K.; Dong, D.; Fang, H.; Levillain, F.; Jin, W.; Mei, J.; Gicquel, B.; Du, Y.; Wang, K.; Gao, Q.; et al. An Interferon-Related Signature in the Transcriptional Core Response of Human Macrophages to Mycobacterium tuberculosis Infection. PLoS ONE 2012, 7, e38367. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Bouchard, G.; Yu, A.; Shafiq, M.; Jamali, M.; Shrager, J.B.; Ayers, K.; Bakr, S.; Gentles, A.J.; Diehn, M.; et al. GFPT2-expressing cancer-associated fibroblasts mediate metabolic reprogramming in human lung adenocarcinoma. Cancer Res. 2018, 78, 3445–3457. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, N.; Liu, Y.; Su, P.; Liang, Y.; Li, Y.; Wang, X.; Chen, T.; Song, X.; Sang, Y.; et al. Epigenetic regulation of NAMPT by NAMPT-AS drives metastatic progression in triple-negative breast cancer. Cancer Res. 2019, 79, 3347–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rienksma, R.A.; Suarez-Diez, M.; Mollenkopf, H.-J.J.; Dolganov, G.M.; Dorhoi, A.; Schoolnik, G.K.; Martins dos Santos, V.; Kaufmann, S.H.E.; Schaap, P.J.; Gengenbacher, M.; et al. Comprehensive insights into transcriptional adaptation of intracellular mycobacteria by microbe-enriched dual RNA sequencing. BMC Genom. 2015, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, R.; Yang, R.; Chen, X.; Harhaj, E.W.; Wang, X.; Fan, Y. Regnase-1, a rapid response ribonuclease regulating inflammation and stress responses. Cell. Mol. Immunol. 2017, 14, 412–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleeson, L.E.; Sheedy, F.J.; Palsson-McDermott, E.M.; Triglia, D.; O’Leary, S.M.; O’Sullivan, M.P.; O’Neill, L.A.J.; Keane, J. Cutting Edge: Mycobacterium tuberculosis Induces Aerobic Glycolysis in Human Alveolar Macrophages That Is Required for Control of Intracellular Bacillary Replication. J. Immunol. 2016, 196, 2444–2449. [Google Scholar] [CrossRef] [Green Version]
- Petruccioli, E.; Scriba, T.J.; Petrone, L.; Hatherill, M.; Cirillo, D.M.; Joosten, S.A.; Ottenhoff, T.H.; Denkinger, C.M.; Goletti, D. Correlates of tuberculosis risk: Predictive biomarkers for progression to active tuberculosis. Eur. Respir. J. 2016, 48, 1751–1763. [Google Scholar] [CrossRef] [Green Version]
- Waddell, S.J.; Stabler, R.A.; Laing, K.; Kremer, L.; Reynolds, R.C.; Besra, G.S. The use of microarray analysis to determine the gene expression profiles of Mycobacterium tuberculosis in response to anti-bacterial compounds. Tuberculosis 2004, 84, 263–274. [Google Scholar] [CrossRef]
- Boshoff, H.I.M.; Myers, T.G.; Copp, B.R.; McNeil, M.R.; Wilson, M.A.; Barry, C.E. The Transcriptional Responses of Mycobacterium tuberculosis to Inhibitors of Metabolism. J. Biol. Chem. 2004, 279, 40174–40184. [Google Scholar] [CrossRef] [Green Version]
- Altaf, M.; Miller, C.H.; Bellows, D.S.; O’Toole, R. Evaluation of the Mycobacterium smegmatis and BCG models for the discovery of Mycobacterium tuberculosis inhibitors. Tuberculosis 2010, 90, 333–337. [Google Scholar] [CrossRef]
- Rampacci, E.; Stefanetti, V.; Passamonti, F.; Henao-Tamayo, M. Preclinical Models of Nontuberculous Mycobacteria Infection for Early Drug Discovery and Vaccine Research. Pathogens 2020, 9, 641. [Google Scholar] [CrossRef]
- Obregón-Henao, A.; Arnett, K.A.; Henao-Tamayo, M.; Massoudi, L.; Creissen, E.; Andries, K.; Lenaerts, A.J.; Ordway, D.J. Susceptibility of mycobacterium abscessus to antimycobacterial drugs in preclinical models. Antimicrob. Agents Chemother. 2015, 59, 6904–6912. [Google Scholar] [CrossRef] [Green Version]
- Macho Rendón, J.; Lang, B.; Ramos Llorens, M.; Gaetano Tartaglia, G.; Torrent Burgas, M. DualSeqDB: The host–pathogen dual RNA sequencing database for infection processes. Nucleic Acids Res. 2021, 49, D687–D693. [Google Scholar] [CrossRef] [PubMed]
- Bosshart, H.; Heinzelmann, M. THP-1 cells as a model for human monocytes. Ann. Transl. Med. 2016, 4, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Bhakta, S.; Kundu, S.; Gupta, M.; Srivastava, B.S.; Srivastava, R. Fast-growing, non-infectious and intracellularly surviving drug-resistant Mycobacterium aurum: A model for high-throughput antituberculosis drug screening. J. Antimicrob. Chemother. 2009, 64, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Arranz-Trullén, J.; Prats-Ejarque, G.; Pulido, D.; Bhakta, S.; Boix, E. Human Antimicrobial RNases Inhibit Intracellular Bacterial Growth and Induce Autophagy in Mycobacteria-Infected Macrophages. Front. Immunol. 2019, 10, 1500. [Google Scholar] [CrossRef]
- Babraham Bioinformatics-FastQC A Quality Control tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 21 August 2018).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Zhai, X.; Xue, Q.; Liu, Q.; Guo, Y.; Chen, Z. Colon cancer recurrence-associated genes revealed by WGCNA co-expression network analysis. Mol. Med. Rep. 2017, 16, 6499–6505. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Stevenson, M.M.; Geary, T.G.; Xia, J. Comprehensive Transcriptome Meta-analysis to Characterize Host Immune Responses in Helminth Infections. PLoS Negl. Trop. Dis. 2016, 10, e0004624. [Google Scholar] [CrossRef]
- Horvath, S. Integrated Weighted Correlation Network Analysis of Mouse Liver Gene Expression Data. In Weighted Network Analysis; Springer: New York, NY, USA, 2011; pp. 321–351. [Google Scholar]
- Botía, J.A.; Vandrovcova, J.; Forabosco, P.; Guelfi, S.; D’Sa, K.; Hardy, J.; Lewis, C.M.; Ryten, M.; Weale, M.E. An additional k-means clustering step improves the biological features of WGCNA gene co-expression networks. BMC Syst. Biol. 2017, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omi. A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological Network Exploration with Cytoscape 3. Curr. Protoc. Bioinforma. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bioproject | Host Cell | Mycobacterium | MOI | POI/h | Ensemble ID | Libraries | Reference 2 |
|---|---|---|---|---|---|---|---|
| PRJNA295153 | THP1 1 | MAB | 10 | 1 h, 4 h, 24 h | 21657 | 27 | [16] |
| PRJNA355844 | BMDM M1 | BCG | 0.18 | 24 h | 25343 | 15 | [19] |
| BMDM M2 | |||||||
| PRJNA471095 | HAM | Mtb-H37Rv | 2 | 2 h, 24 h, 72 h | 20774 | 36 | [20] |
| BMDM | |||||||
| PRJNA495462 | BMDM | Mtb-H37Rv | 0.5 | 18 h | 39354 | 6 | [21] |
| PRJNA575195 | THP1 | M. aurum | 10 | 24 h | 58233 | 6 | |
| PRJNA279959 | BMDM | Mtb-H37Rv | 2 | 4 h, 18 h, 48 h | 12728 | 108 | [13] |
| H37Rv + | |||||||
| M. smegmatis | |||||||
| Mtb-GC1237 | |||||||
| BCG |
| Strain | Growth Rate | Pathogenicity | Morphology | Note |
|---|---|---|---|---|
| Mycobacterium tuberculosis Mtb H37Rv | Slow growth | Tuberculosis | Rough | Laboratory strain |
| Mycobacterium tuberculosis GC1237 | Slow growth | Tuberculosis | Rough | |
| M. bovis Bacillus Calmette–Guérin (BCG) | Slow growth | Non-pathogenic | Rough | An attenuated strain of M. bovis, used as a vaccine for TB |
| M. smegmatis | Fast growth | Non-pathogenic | Rough | |
| M. abscessus Rough (ABR) | Fast growth | Pathogenic | Rough | |
| M. abscessus Smooth (ABS) | Fast growth | Pathogenic | Smooth | |
| M. aurum | Fast growth | Non-pathogenic | Smooth |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, L.; Wei, R.; Bhakta, S.; Waddell, S.J.; Boix, E. Weighted Gene Co-Expression Network Analysis Identifies Key Modules and Hub Genes Associated with Mycobacterial Infection of Human Macrophages. Antibiotics 2021, 10, 97. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020097
Lu L, Wei R, Bhakta S, Waddell SJ, Boix E. Weighted Gene Co-Expression Network Analysis Identifies Key Modules and Hub Genes Associated with Mycobacterial Infection of Human Macrophages. Antibiotics. 2021; 10(2):97. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020097
Chicago/Turabian StyleLu, Lu, RanLei Wei, Sanjib Bhakta, Simon J. Waddell, and Ester Boix. 2021. "Weighted Gene Co-Expression Network Analysis Identifies Key Modules and Hub Genes Associated with Mycobacterial Infection of Human Macrophages" Antibiotics 10, no. 2: 97. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020097