
Mechanistic Fingerprinting Reveals Kinetic Signatures of Resistance to Daptomycin and Host Defense Peptides in Streptococcus mitis-oralis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. S. mitis-oralis Strains
2.2. Host Defense Peptides (HDPs)
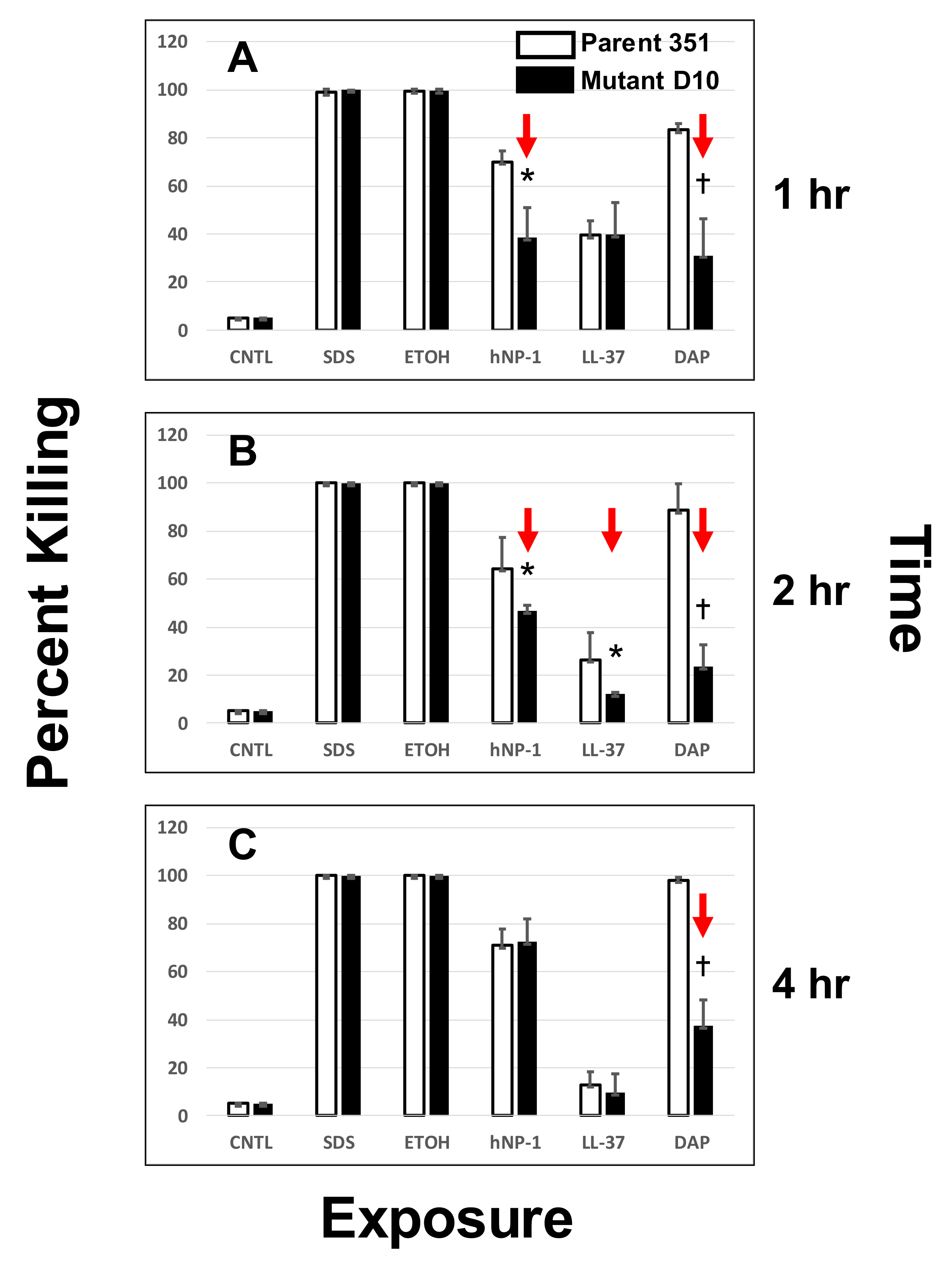
2.3. Susceptibility to DAP and HDPs by Time-Kill Assays
2.4. BODIPY-DAP Binding
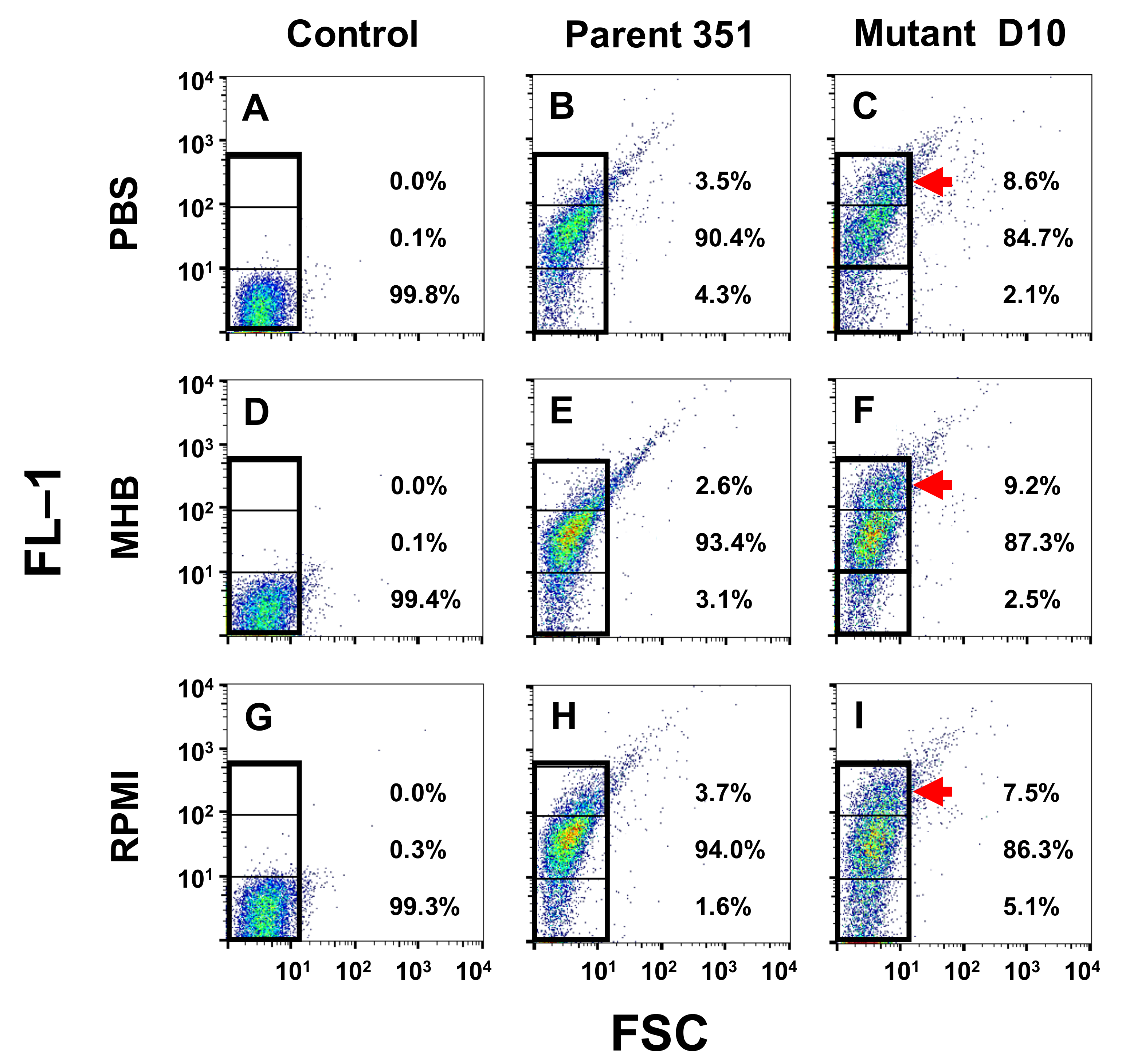
2.5. Kinetic Signatures by Multi-Parameter Flow Cytometry
2.6. Statistical Analysis
3. Results
3.1. BODIPY-DAP Binding Interactions
3.2. DAP- or HDP-Mediated Timed-Killing
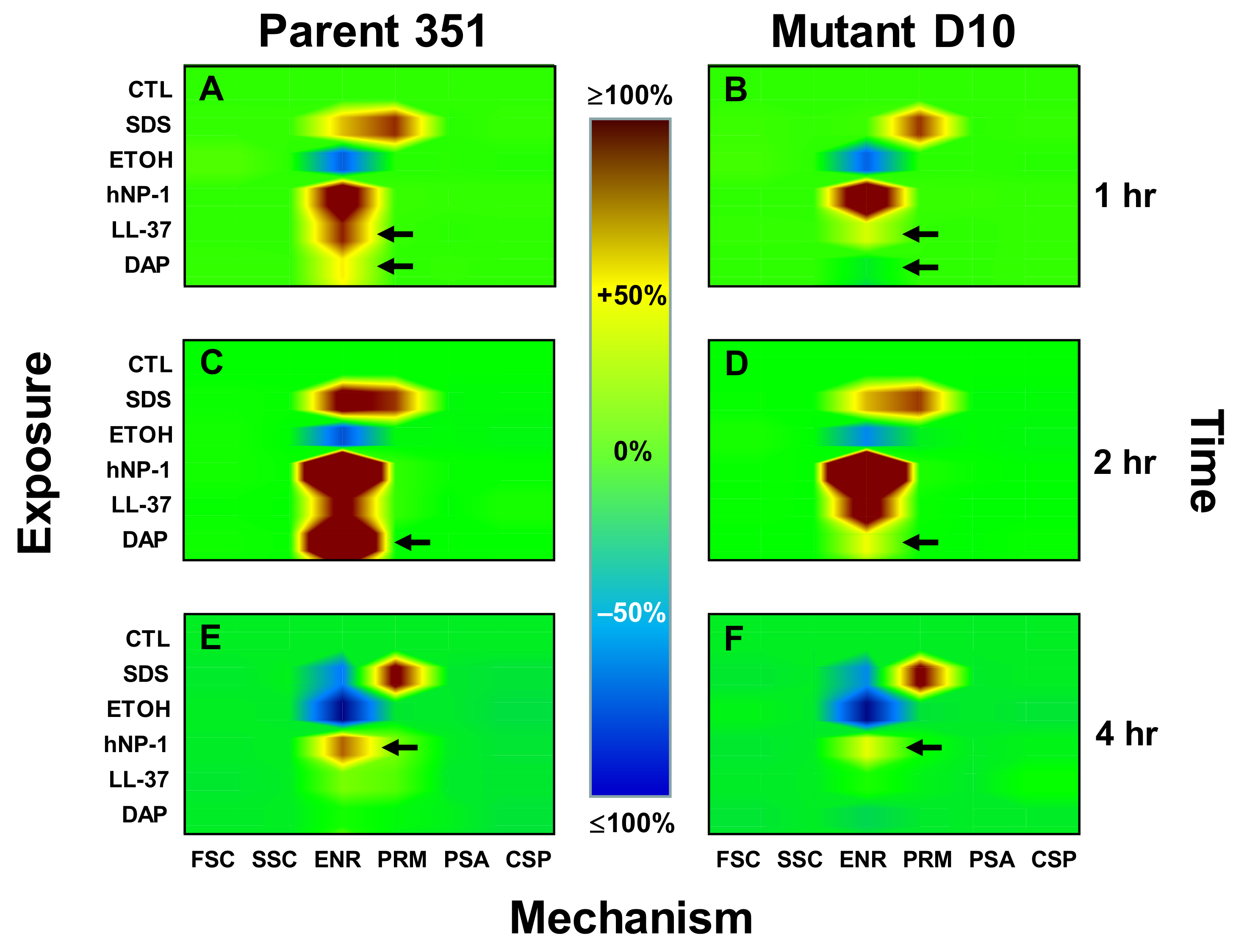
3.3. Kinetic Signatures by Multi-Parameter Flow Cytometry
3.3.1. One-Hour Exposure Time Point
3.3.2. Two-Hour Exposure Time Point
3.3.3. Four-Hour Exposure Time Point
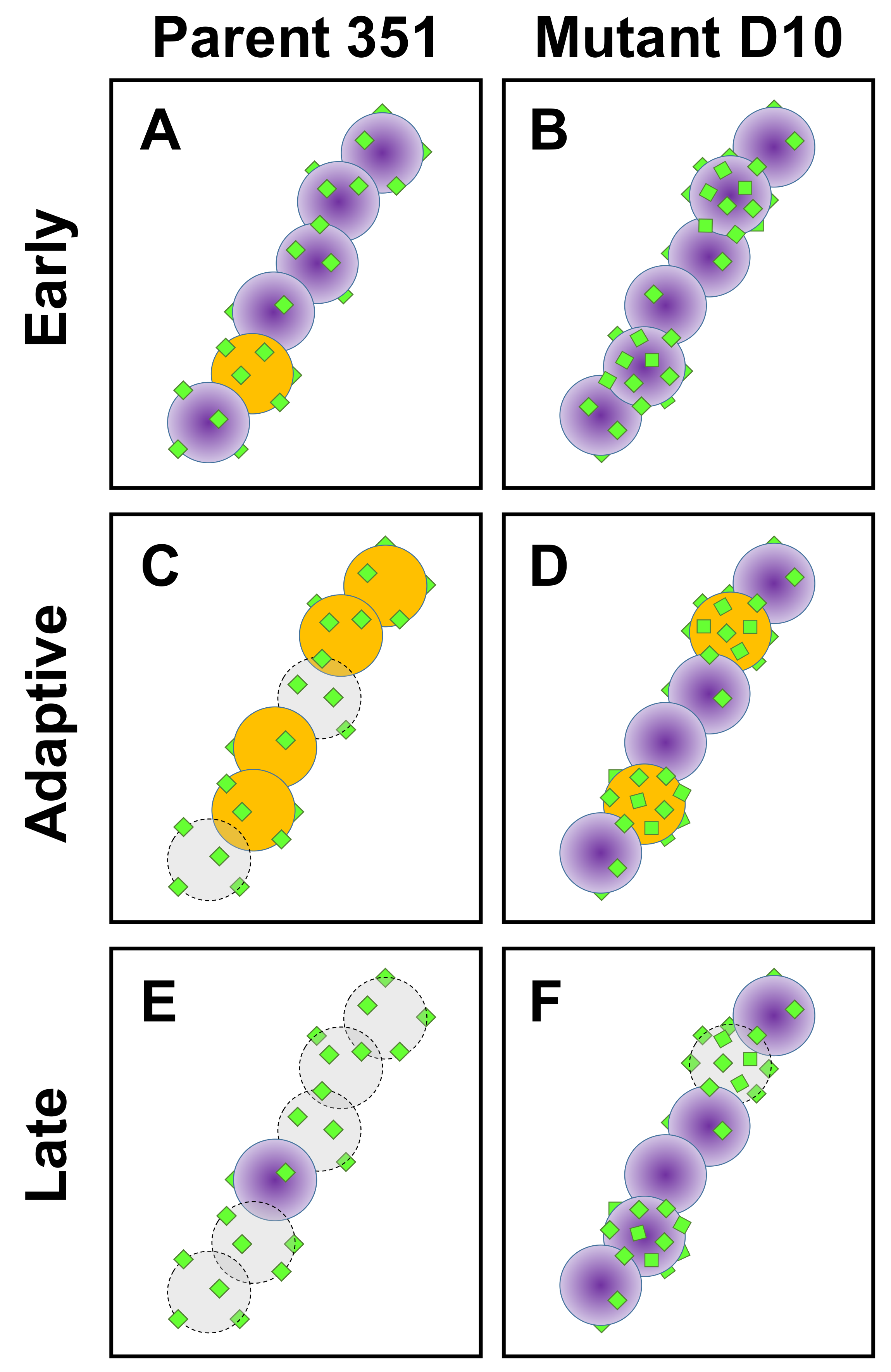
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holland, T.L.; Bayer, A.S.; Fowler, V.G. Endocarditis and Intravascular Infections. In Principles and Practices of Infectious Diseases, 9th ed.; Mandell, G.L., Bennett, J.E., Eds.; Elsevier: Philadelphia, PA, USA, 2020; Chapter 80. [Google Scholar]
- Ahmed, R.; Hassall, T.; Morland, B.; Gray, J. Viridans streptococcus bacteremia in children on chemotherapy for cancer: An underestimated problem. Pediatr. Hematol. Oncol. 2003, 20, 439–444. [Google Scholar] [CrossRef]
- Husain, E.; Whitehead, S.; Castell, A.; Thomas, E.E.; Speert, D.P. Viridans streptococci bacteremia in children with malignancy: Relevance of species identification and penicillin susceptibility. Pediatr. Infect. Dis. J. 2005, 24, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Marron, A.; Carratala, J.; Gonzalez-Barca, E.; Fernandez-Sevilla, A.; Alcaide, F.; Gudiol, F. Serious complications of bacteremia caused by Viridans streptococci in neutropenic patients with cancer. Clin. Infect. Dis. 2000, 31, 1126–1130. [Google Scholar] [CrossRef]
- Huang, W.-T.; Chang, L.-Y.; Hsueh, P.-R.; Lu, C.-Y.; Shao, P.-L.; Huang, F.-Y.; Lee, P.-I.; Chen, C.-M.; Lee, C.-Y.; Huang, L.-M. Clinical features and complications of viridans streptococci bloodstream infection in pediatric hemato-oncology patients. J. Microbiol. Immunol. Infect. 2007, 40, 349–354. [Google Scholar] [PubMed]
- Shelburne, S.A.; Sahasrabhojane, P.; Saldana, M.; Yao, H.; Su, X.; Horstmann, N.; Thompson, E.; Flores, A.R. Streptococcus mitis trains causing severe clinical disease in cancer patients. Emerg. Infect. Dis. 2014, 20, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Freifeld, A.G.; Razonable, R.R. Viridans group streptococci in febrile neutropenic cancer patients: What should we fear? Clin. Infect. Dis. 2014, 59, 231–233. [Google Scholar] [CrossRef]
- Prabhu, R.M.; Piper, K.E.; Baddour, L.M.; Steckelberg, J.M.; Wilson, W.R.; Patel, R. Antimicrobial susceptibility patterns among viridans group streptococci isolates from infective endocarditis patients from 1971–1986 and 1994 to–2002. Antimicrob. Agents Chemother. 2004, 48, 4463–4465. [Google Scholar] [CrossRef] [Green Version]
- Shelburne, S.A.; Lasky, R.E.; Sahasrabhojane, P.; Tarrand, J.T.; Rolston, K.V.I. Development and validation of a clinical model to predict the presence of β-lactam resistance in viridians group streptococci causing bacteremia in neutropenic cancer patients. Clin. Infect. Dis. 2014, 59, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Ron-Bin, H.; Lin, F.-Y. Effect of penicillin resistance on presentation and outcome of nonenterococcal streptococcal infective endocarditis. Cardiology 2006, 105, 234–239. [Google Scholar]
- Sabella, C.; Murphy, D.; Drummond-Webb, J. Endocarditis due to Streptococcus mitis with high-level resistance to penicillin and ceftriaxone. JAMA 2001, 285, 2195. [Google Scholar] [CrossRef]
- Saldar, A.; Rolston, K.V. Vancomycin tolerance a potential mechanism for refractory gram-positive bacteria: Observational study in patients with cancer. Cancer 2006, 106, 1815–1820. [Google Scholar]
- Garcia-de-la-Maria, C.; Pericas, J.M.; del Rio, A.; Castañeda, X.; Vila-Farrés, X.; Armero, Y.; Espinal, P.A.; Cervera, C.; Soy, D.; Falces, C.; et al. Early in Vitro and in Vivo development of high-level daptomycin resistance is common in mitis group of streptococci after exposure to daptomycin. Antimicrob. Agents Chemother. 2013, 57, 2319–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, J.; Smith, J.R.; Singh, N.B.; Rice, S.; Stamper, K.; De La Maria, C.G.; Bayer, A.S.; Mishra, N.N.; Miró, J.M.; Tran, T.T.; et al. Evaluation of daptomycin combinations with cephalosporins or gentamicin against Streptococcus mitis group strains in an In Vitro model of simulated endocardial vegetations (SEVs). J. Antimicrob. Chemother. 2017, 72, 2290–2296. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.S.; Schneider, T.; Sahl, H.-G. Mechanisms of daptomycin resistance in Staphylococcus aureus: Role of the cell membrane and cell wall. Ann. N. Y. Acad. Sci. 2013, 1277, 139–158. [Google Scholar] [CrossRef] [Green Version]
- Kaatz, G.W.; Lundstrom, T.S.; Seo, S.M. Mechanisms of daptomycin resistance in Staphylococcus aureus. Int. J. Antimicrob. Agents 2006, 28, 280–287. [Google Scholar]
- Arias, C.A.; Panesso, D.; McGrath, D.M.; Qin, X.; Mojica, M.F.; Miller, C.; Diaz, L.; Tran, T.T.; Rincon, S.; Barbu, E.M.; et al. Genetic basis for in vivo daptomycin resistance in enterococci. N. Engl. J. Med. 2011, 365, 892–900. [Google Scholar]
- Tran, T.T.; Mishra, N.N.; Seepersaud, R.; Diaz, L.; Rios, R.; Dinh, A.Q.; Garcia-De-La-Maria, C.; Rybak, M.J.; Miro, J.M.; Shel-burne, S.A.; et al. Failure of high-dose daptomycin for bacteremia caused by daptomycin-susceptible Enterococcus faecium harboring LiaSR substitutions. Clin. Infect. Dis. 2014, 59, 1277–1280. [Google Scholar]
- Mishra, N.N.; Tran, T.T.; Seepersaud, R.; Garcia-de-la-Maria, C.; Faull, K.; Yoon, A.; Miro, J.M.; Rybak, M.J.; Bayer, A.S.; Arias, C.A.; et al. Perturbations of phosphatidate cytidylyltransferase (CdsA) mediate daptomycin resistance in Streptococcus mitis by a novel mechasnism. Antimicrob. Agents Chemother. 2017, 61, e02435-16. [Google Scholar] [CrossRef] [Green Version]
- Adams, H.M.; Joyce, L.R.; Guan, Z.; Akins, R.L.; Palmer, K.L. Streptococcus mitis and Streptococcus oralis mutate an ‘essential’ gene upon exposure to daptomycin. FASEB J. 2018. [Google Scholar] [CrossRef]
- Tran, T.T.; Mishra, N.N.; Seepersaud, R.; Diaz, L.; Rios, R.; Dinh, A.Q.; Garcia-de-la-Maria, C.; Rybak, M.J.; Miro, J.M.; Shelburne, S.; et al. Mutations in cdsA and pgsA correlate with daptomycin resistance in Streptococcus mitis and S. oralis. Antimicrob. Agents Chemother. 2018, 63, e01531-18. [Google Scholar]
- Parrett, A.; Reed, J.M.; Gardner, S.G.; Mishra, N.N.; Bayer, A.S.; Powers, R.; Somerville, G.A. Metabolic changes associated with adaptive resistance to daptomycin in Streptococcus mitis-oralis. BMC Microbiol. 2020, 20, 162. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Approved standard M100-SCLSI; CLSI: Wayne, PA, USA, 2020. [Google Scholar]
- Ganz, T.; Selsted, M.E.; Szklarek, D.; Harwig, S.S.; Daher, K.; Bainton, D.F.; Lehrer, R.I. Defensins: Natural peptide antibiotics of human neutrophils. J. Clin. Investig. 1985, 76, 1427–1435. [Google Scholar]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-Like Receptor Triggering of a Vitamin D-Mediated Human Antimicrobial Response. Science 2006, 311, 1770–1773. [Google Scholar]
- Mishra, N.N.; McKinnell, J.; Yeaman, M.R.; Rubio, A.; Nast, C.C.; Chen, L.; Kreiswirth, B.N.; Bayer, A.S. In VitroCross-Resistance to Daptomycin and Host Defense Cationic Antimicrobial Peptides in Clinical Methicillin-Resistant Staphylococcus aureus Isolates. Antimicrob. Agents Chemother. 2011, 55, 4012–4018. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Dietz, M.J.; Li, B. Antimicrobial peptide LL-37 is bactericidal against Staphylococcus aureus biofilms. PLoS ONE 2019, 14, e0216676. [Google Scholar]
- Chaili, S.; Cheung, A.L.; Bayer, A.S.; Xiong, Y.Q.; Waring, A.J.; Memmi, G.; Donegan, N.; Yang, S.J.; Yeaman, M.R. The GraS sensor in Staphylococcus aureus mediates resistance to host defense peptides differing in mechanisms of action. Infect. Immun. 2015, 84, 459–466. [Google Scholar]
- Mishra, N.N.; Tran, T.T.; Arias, C.A.; Seepersaud, R.; Sullam, P.M.; Bayer, A.S. Strain-Specific Adaptations of Streptococcus mitis-oralis to Serial In Vitro Passage in Daptomycin (DAP): Genotypic and Phenotypic Characteristics. Antibiotics 2020, 9, 520. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.S.; Lasken, R.S. Single cell genomics of bacterial pathogens: Outlook for infectious disease research. Genome Med. 2014, 6, 108. [Google Scholar] [CrossRef] [Green Version]
- Garcia-De-La-Maria, C.; Xiong, Y.Q.; Pericas, J.M.; Armero, Y.; Moreno, A.; Mishra, N.N.; Rybak, M.J.; Tran, T.T.; Arias, C.A.; Sullam, P.M.; et al. Impact of High-Level Daptomycin Resistance in the Streptococcus mitis Group on Virulence and Survivability during Daptomycin Treatment in Experimental Infective Endocarditis. Antimicrob. Agents Chemother. 2017, 61, e02418-16. [Google Scholar]
- Zapata, B.; Alvarez, D.N.; Farah, S.; Garcia-De-La-Maria, C.; Miro, J.M.; Sakoulas, G.; Bayer, A.S.; Mishra, N.N. Prevention of High-Level Daptomycin-Resistance Emergence In Vitro in Streptococcus mitis-oralis by Using Combination Antimicrobial Strategies. Curr. Microbiol. 2018, 75, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Munita, J.M.; Arias, C.A. Mechanisms of drug resistance: Daptomycin resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 32–53. [Google Scholar]
- Kang, Y.; McMillan, I.; Norris, M.H.; Hoang, T.T. Single prokaryotic cell isolation and total transcript amplification protocol for transcriptomic analysis. Nat. Protoc. 2015, 10, 974–984. [Google Scholar]
- Marston, A.L.; Thomaides, H.B.; Edwards, D.H.; Sharpe, M.E.; Errington, J. Polar localization of the MinD protein of Bacillus subtilis and its role in selection of the mid-cell division site. Genes Dev. 1998, 12, 3419–3430. [Google Scholar] [CrossRef] [Green Version]
- Strahl, H.; Hamoen, L.W. Membrane potential is important for bacterial cell division. Proc. Natl. Acad. Sci. USA 2010, 107, 12281–12286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, A.; Wenzel, M.; Strahl, H.; Grein, F.; Saaki, T.N.V.; Kohl, B.; Siersma, T.; Bandow, J.E.; Sahl, H.-G.; Schneider, T.; et al. Daptomycin inhibits cell envelope synthesis by interfering with fluid membrane microdomains. Proc. Natl. Acad. Sci. USA 2016, 113, E7077–E7086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, A.; Wenzel, M.; Strahl, H.; Grein, F.; Saaki, T.N.; Kohl, B.; Siersma, T.; Bandow, J.E.; Sahl, H.G.; Schneider, T.; et al. Staphylococcus aureus metabolic adaptations during the transition from a daptomycin susceptibility phenotype to a daptomycin nonsusceptibility phenotype. Antimicrob. Agents Chemother. 2015, 59, 4226–4238. [Google Scholar]
- Yeaman, M.R.; Bayer, A.S.; Koo, S.P.; Foss, W.; Sullam, P.M. Platelet microbicidal proteins and neutrophil defensin disrupt the Staphylococcus aureus cytoplasmic membrane by distinct mechanisms of action. J. Clin. Investig. 1998, 101, 178–187. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeaman, M.R.; Chan, L.C.; Mishra, N.N.; Bayer, A.S. Mechanistic Fingerprinting Reveals Kinetic Signatures of Resistance to Daptomycin and Host Defense Peptides in Streptococcus mitis-oralis. Antibiotics 2021, 10, 404. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10040404
Yeaman MR, Chan LC, Mishra NN, Bayer AS. Mechanistic Fingerprinting Reveals Kinetic Signatures of Resistance to Daptomycin and Host Defense Peptides in Streptococcus mitis-oralis. Antibiotics. 2021; 10(4):404. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10040404
Chicago/Turabian StyleYeaman, Michael R., Liana C. Chan, Nagendra N. Mishra, and Arnold S. Bayer. 2021. "Mechanistic Fingerprinting Reveals Kinetic Signatures of Resistance to Daptomycin and Host Defense Peptides in Streptococcus mitis-oralis" Antibiotics 10, no. 4: 404. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10040404