
Staphylococcus aureus RnpA Inhibitors: Computational-Guided Design, Synthesis and Initial Biological Evaluation

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
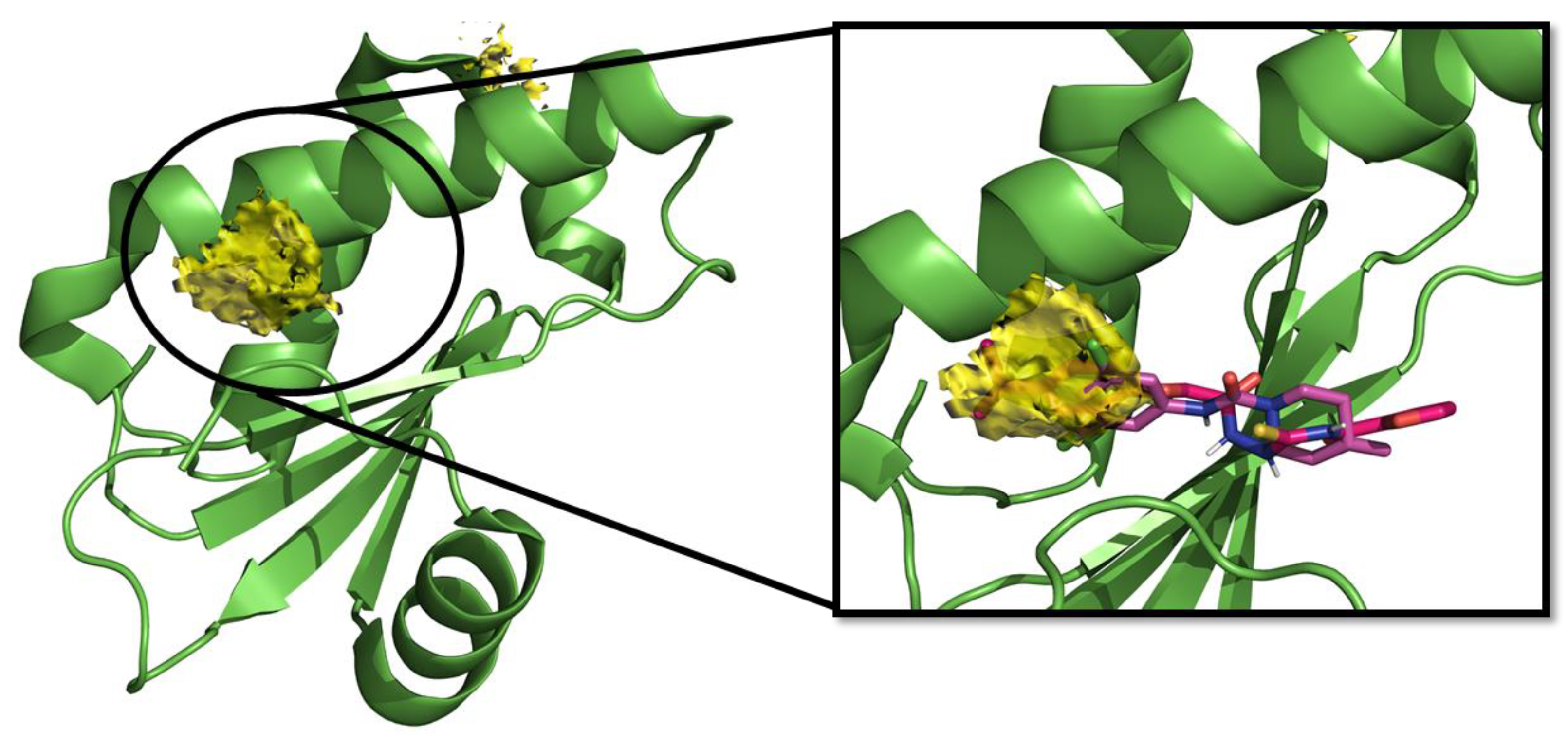
2.1. Computational Studies and Design
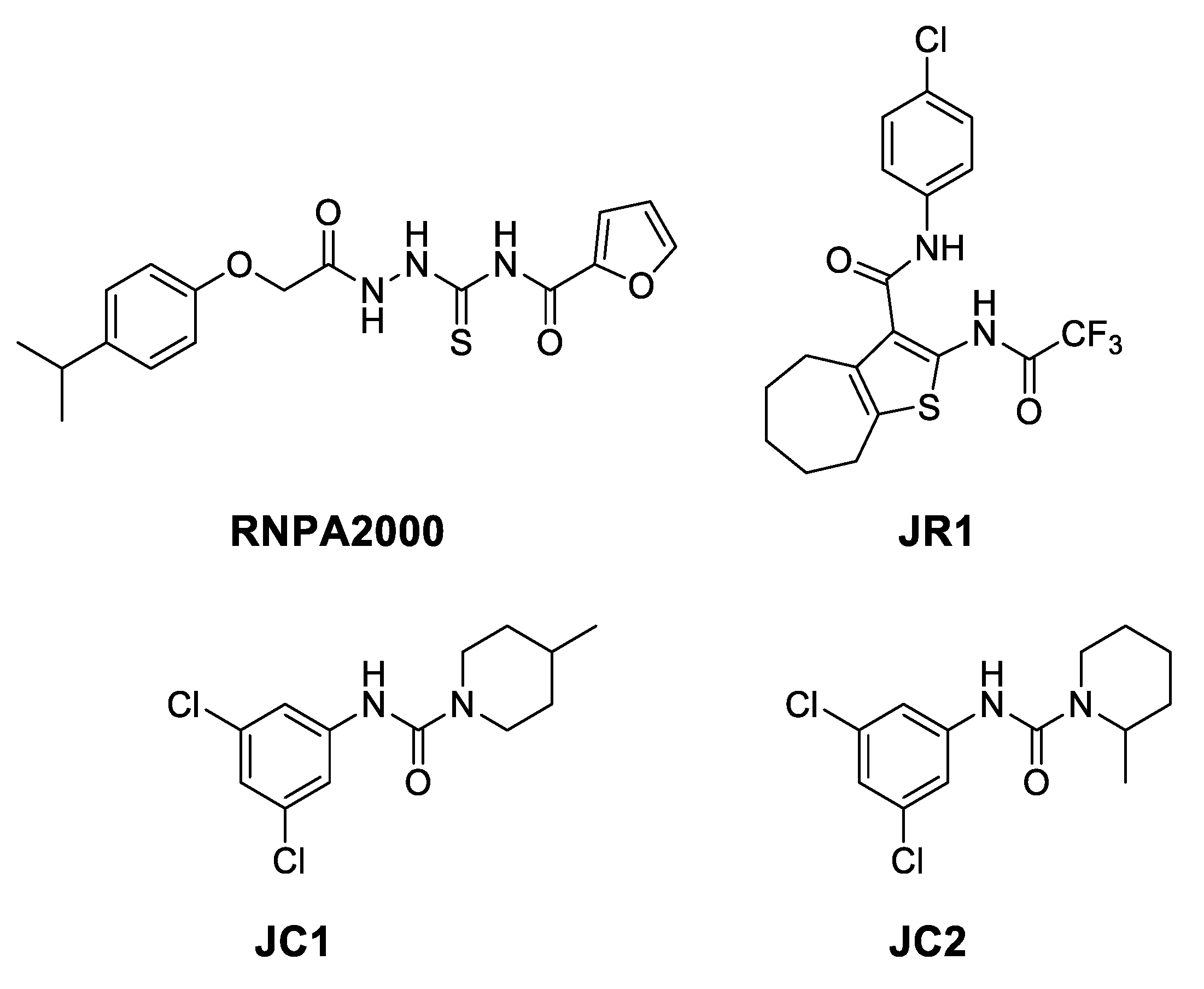
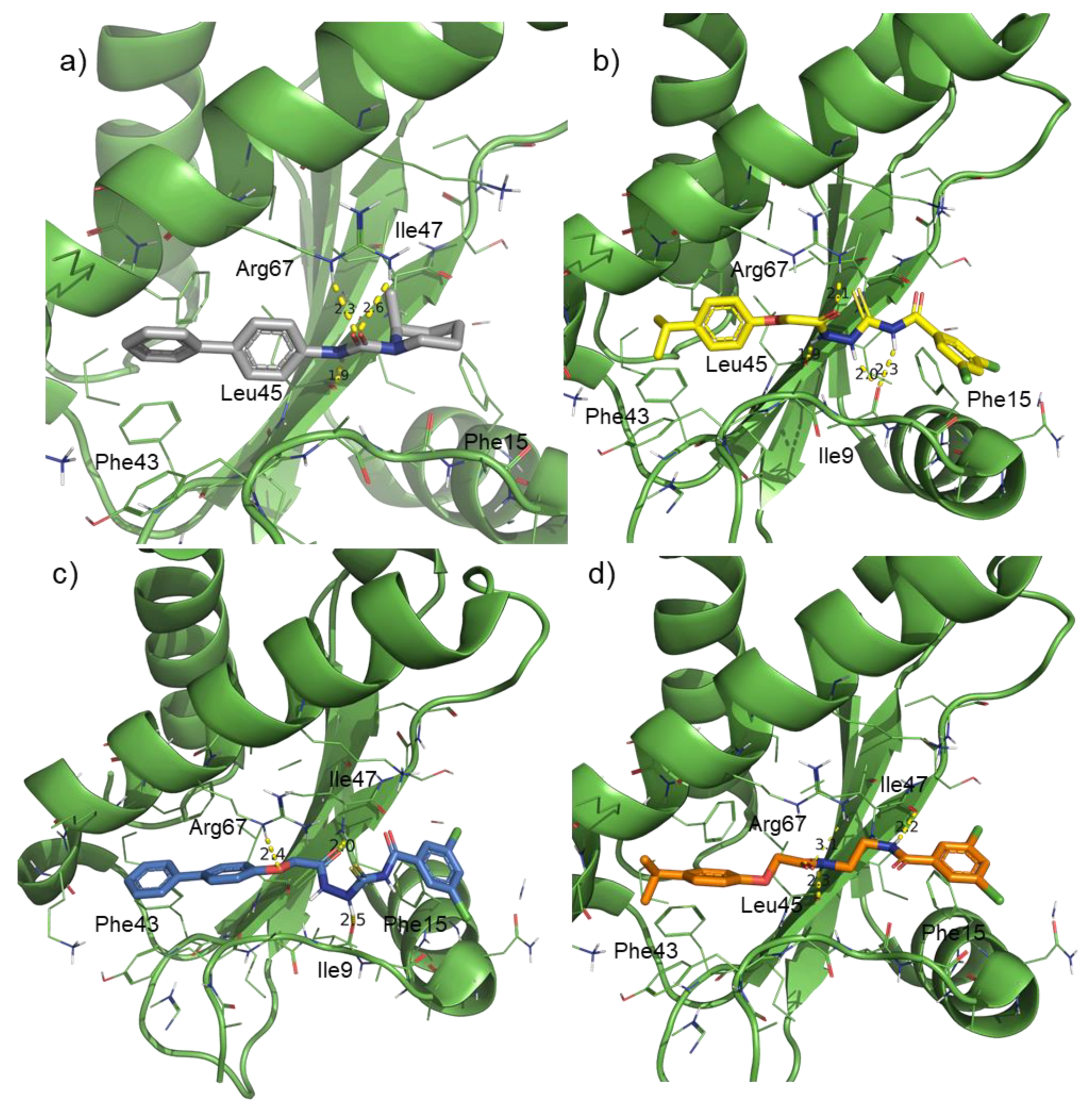
- in the subregion where the main hydrophobic hotspot was identified, there are several aromatic and hydrophobic residues, including Tyr7, Phe43, Leu45, and Phe70, where the first two are generating a cleft. This might help in anchoring aromatic systems, establishing π or CH–π interactions (such as the i-propylphenyl group that is present in RnpA2000);
- the central part contains a solvent-exposed area, which might allow the binding of linear linkers by establishing hydrogen bonds interactions via the backbone of Ile9, Leu45, Ile47 or the guanidinic moiety of Arg67. H-bonds donor-acceptor that are predicted to interact with the ureidic portion of JC1 and with the semi-thiocarbazide moiety of RNPA2000; and,
- finally, at the other end, Phe15 and Lys63 are suitable for π interaction or π-cation interactions with different aromatic moieties, such as the one present in RNA2000.
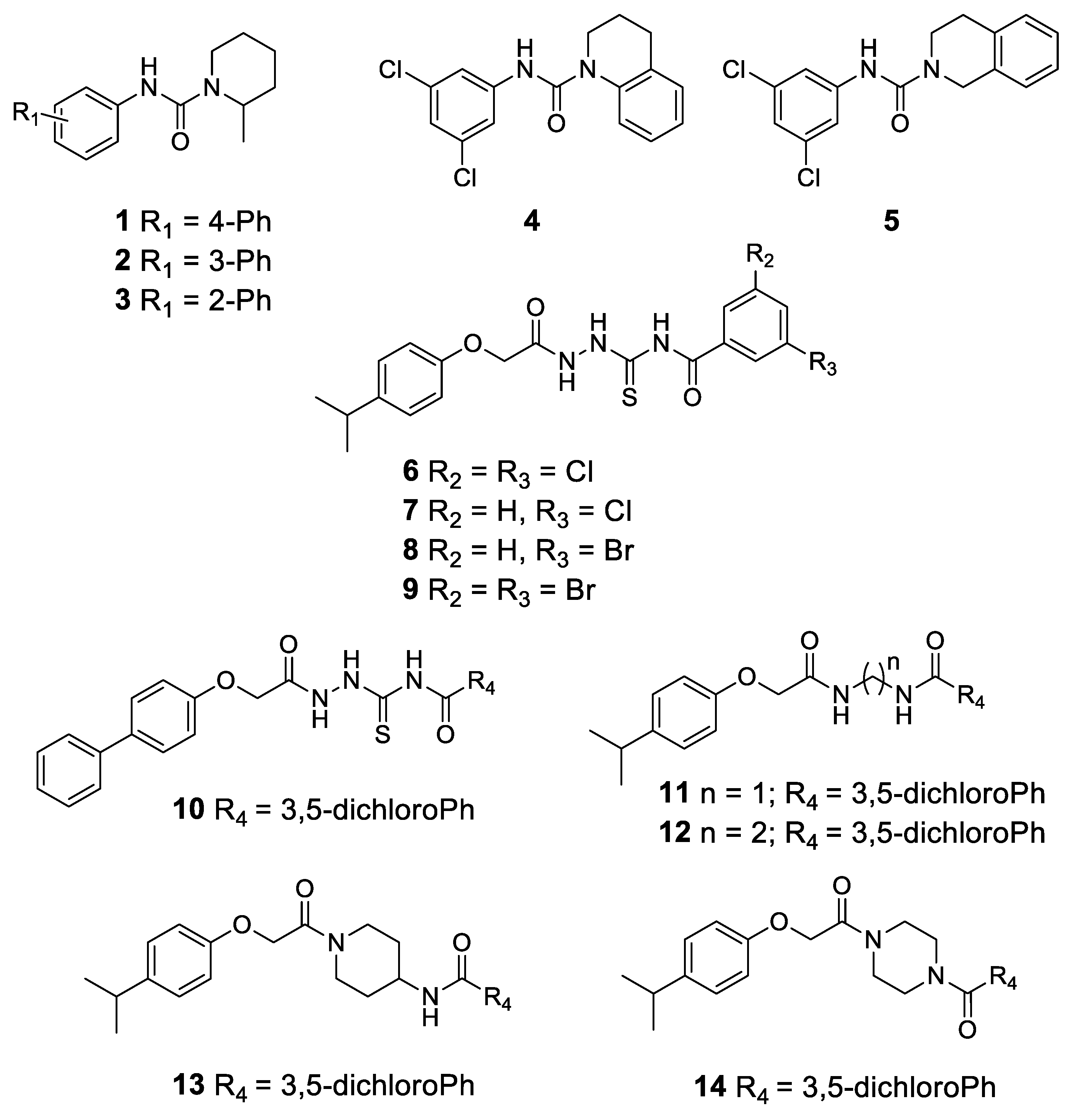
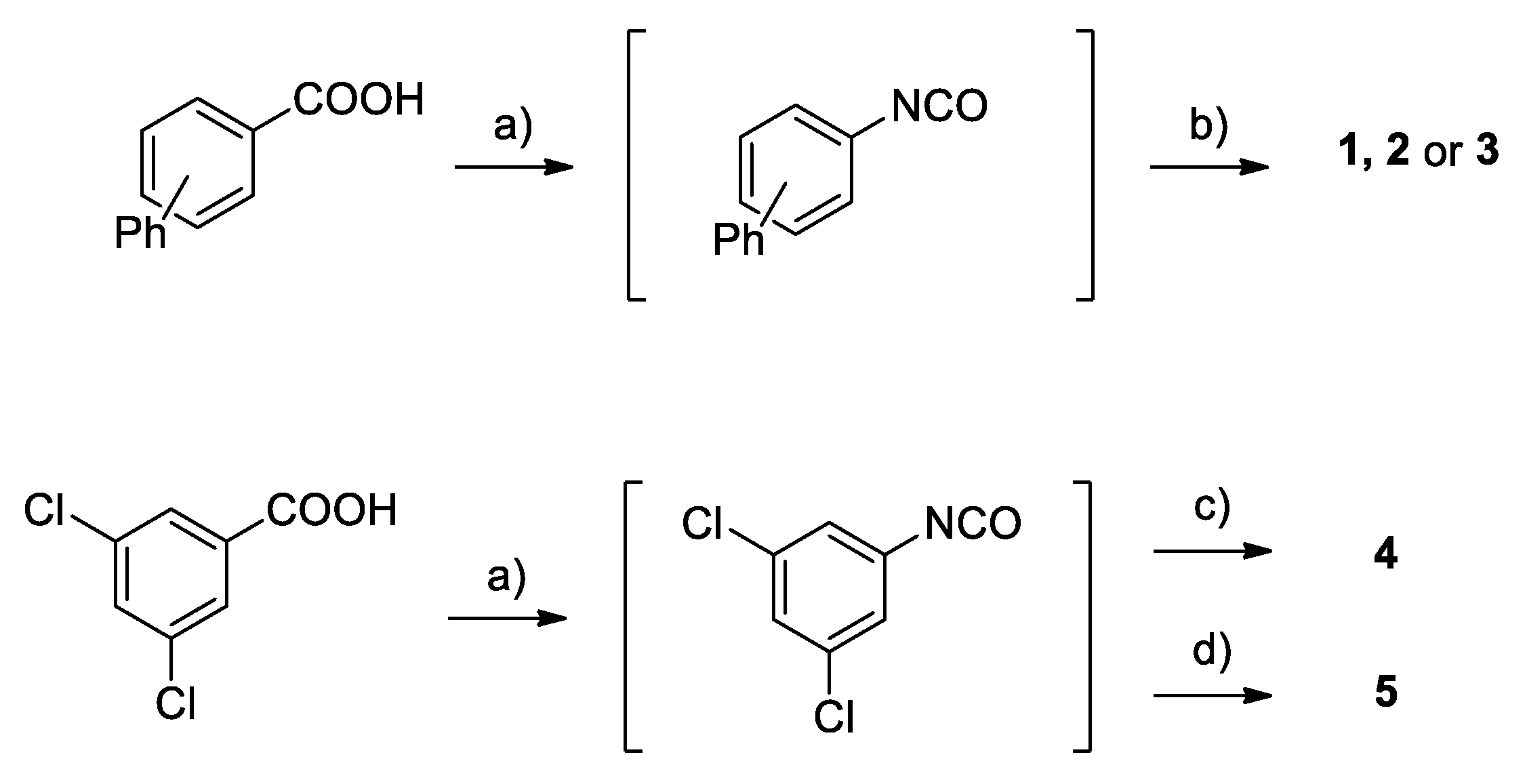
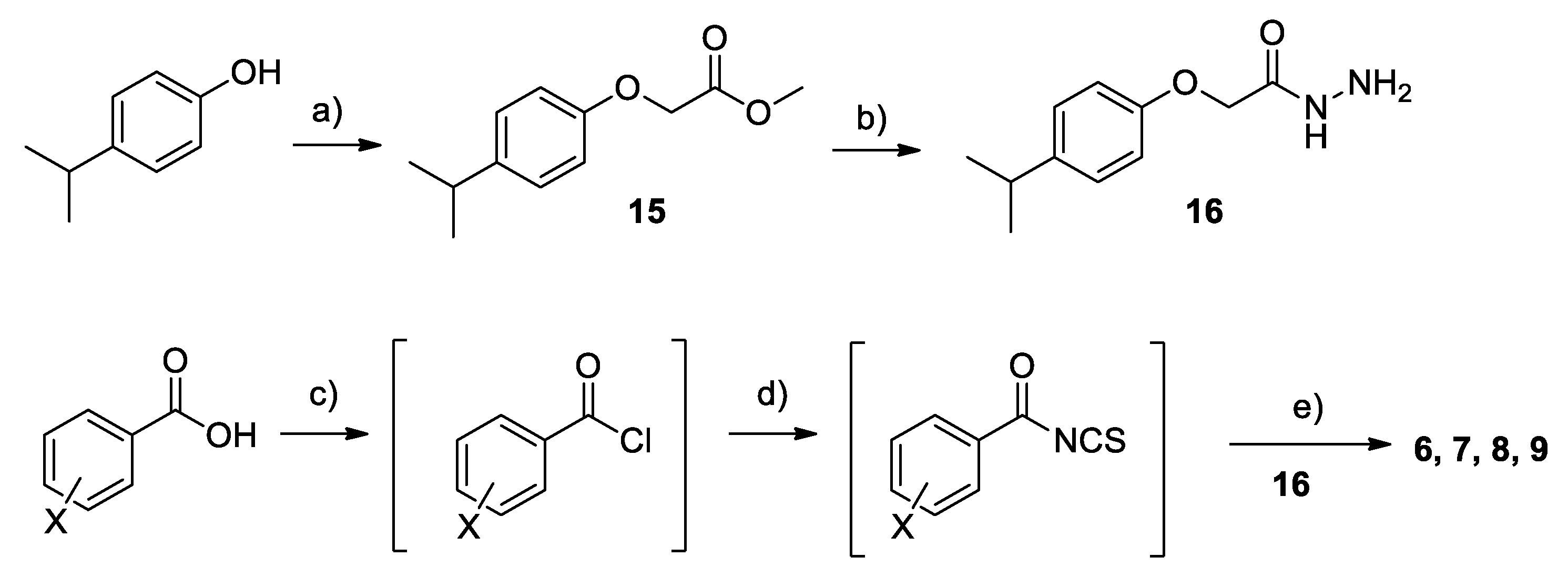
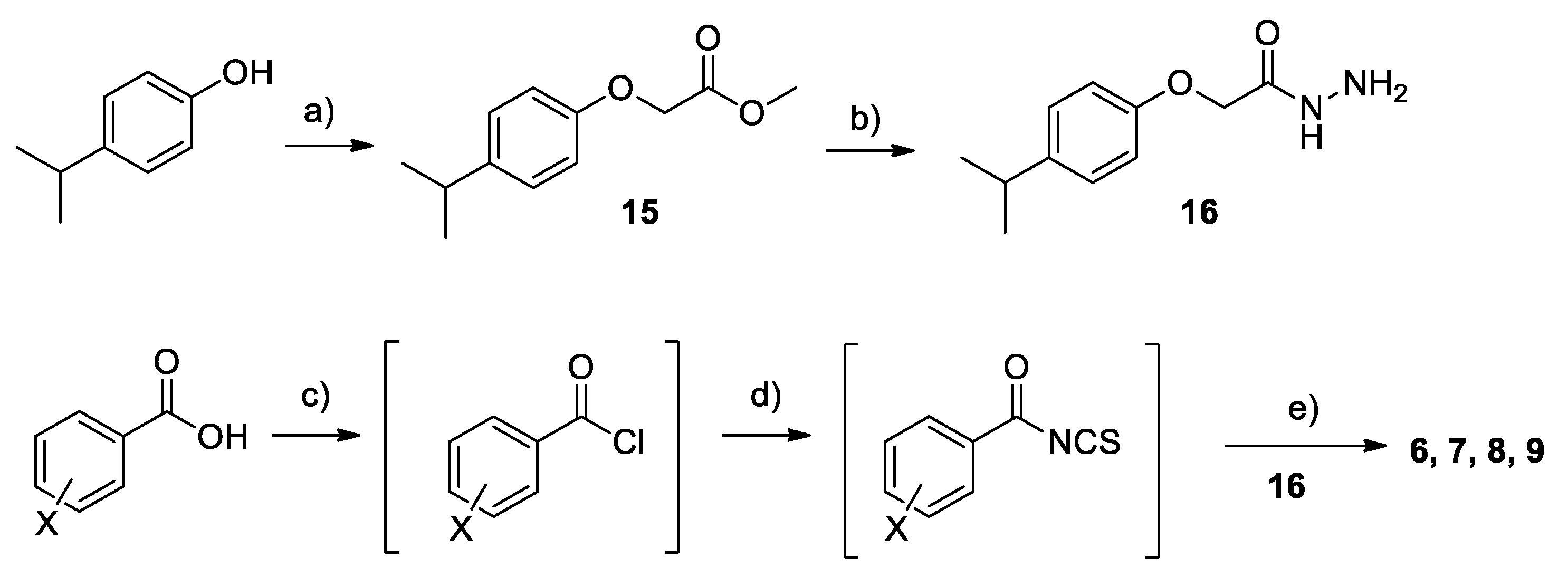
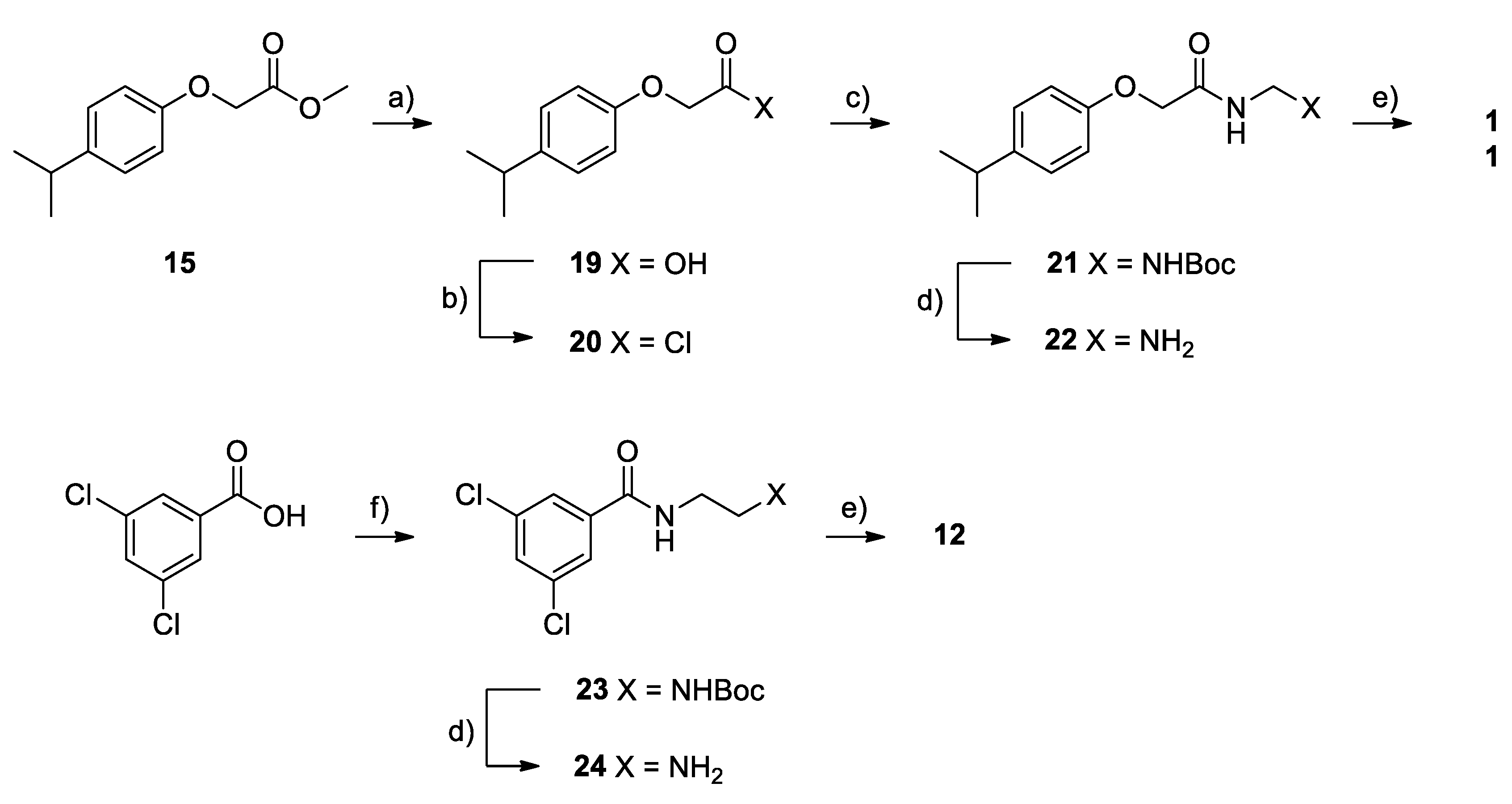
2.2. Chemistry
2.3. Biological Evaluation
2.3.1. Antimicrobial Activity
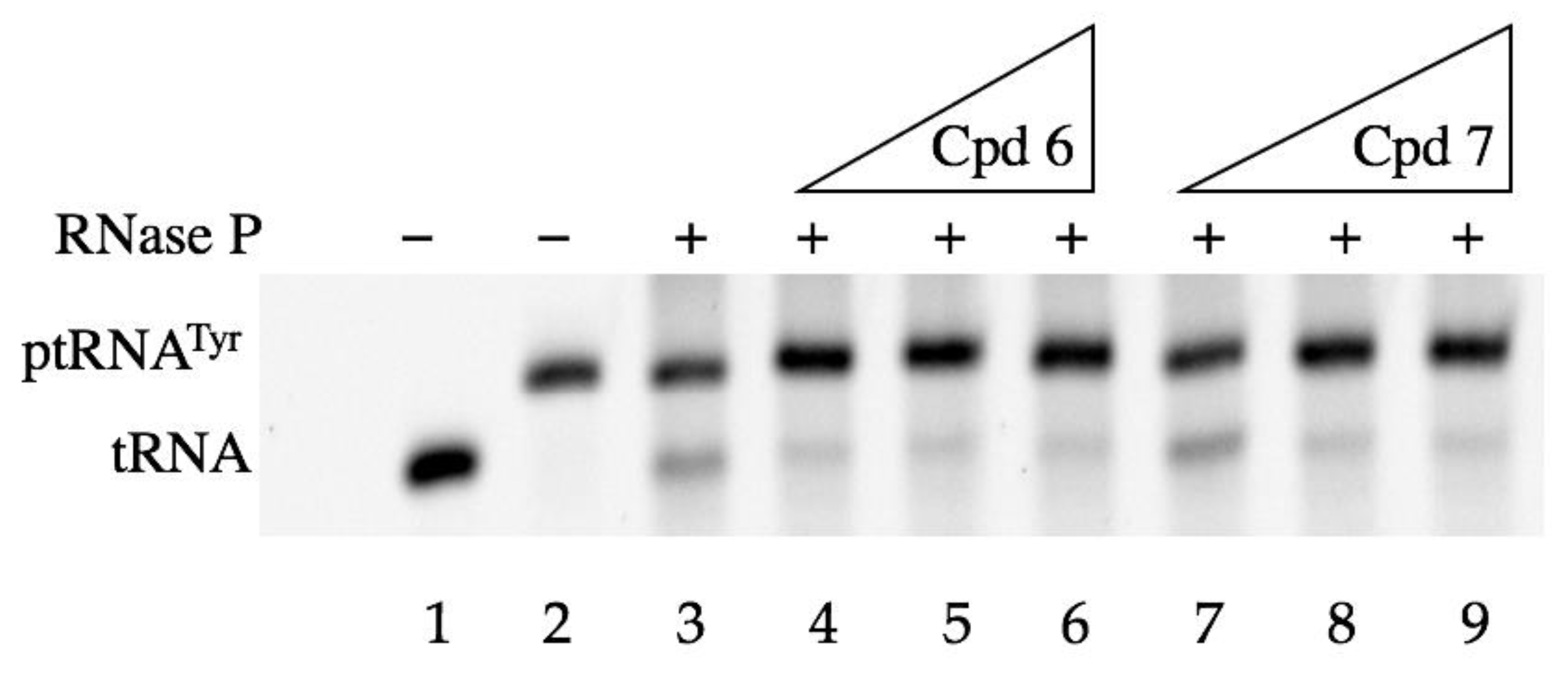
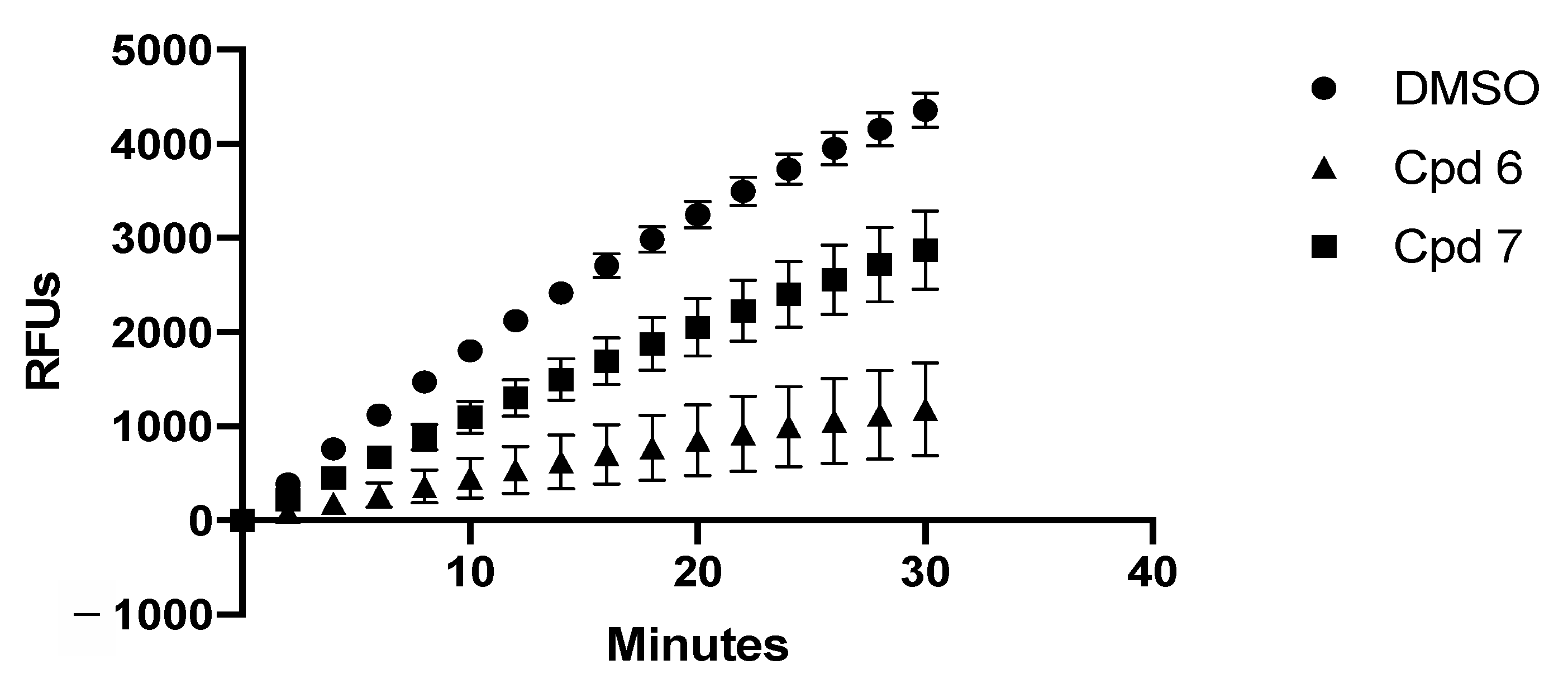
2.3.2. In-Vitro Assays
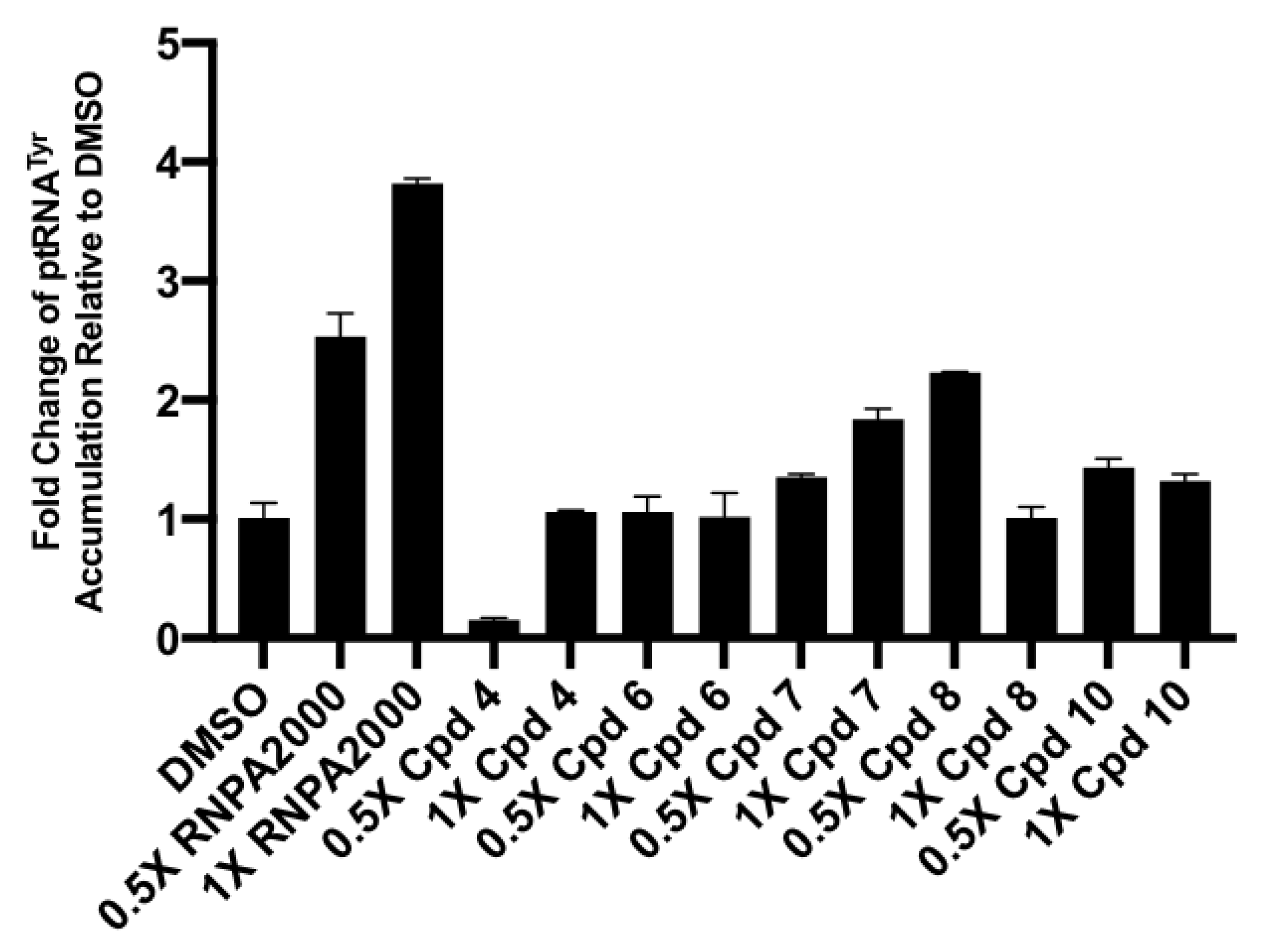
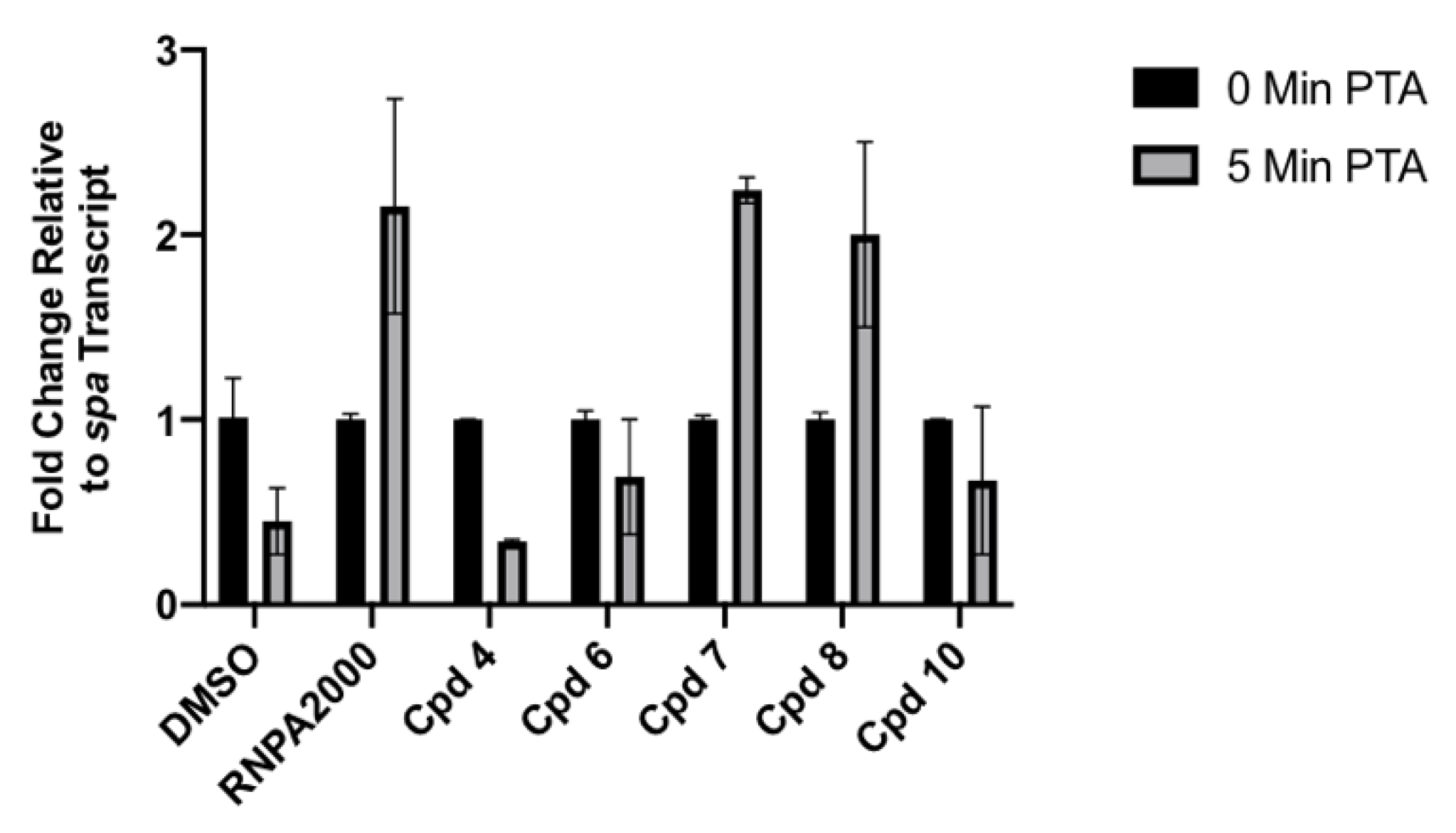
2.3.3. Cellular Assays
3. Conclusions
4. Materials and Methods
4.1. Chemistry
Synthesis
4.2. Biological Evaluation
4.2.1. Bacterial Growth Conditions
4.2.2. Antimicrobial Susceptibility Testing
ATCC 29213 and ATCC 43300
4.2.3. RnpA Protein Purification
4.2.4. In-Vitro Transcription of RNA
4.2.5. In-Vitro ptRNA Processing Assays
4.2.6. In-Vitro mRNA Degradation Assays
4.2.7. Cellular tRNATyr Population Measures
4.2.8. Cellular mRNA Turnover Assays
4.2.9. Bacterial RNA Isolation and Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.3. Hotspot Maps
4.4. Computational Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Dutescu, I.A.; Hillier, S.A. Encouraging the Development of New Antibiotics: Are Financial Incentives the Right Way Forward? A Systematic Review and Case Study. Infect. Drug Resist. 2021, 14, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Global Action Plan. Available online: https://www.who.int/antimicrobial-resistance/publications/global-action-plan/en/ (accessed on 12 February 2021).
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance#:~:text=What%20is%20antimicrobial%20resistance%3F,spread%2C%20severe%20illness%20and%20death (accessed on 12 February 2021).
- CDC. Antibiotic Resistance Threats in the United States; Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019.
- Aminov, R.I. A brief history of the antibiotic era: Lessons learned and challenges for the future. Front. Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, C.; Brooks, L.; Beckley, A.; Colquhoun, J.; Dewhurst, S.; Dunman, P.M. Neomycin Sulfate Improves the Antimicrobial Activity of Mupirocin-Based Antibacterial Ointments. Antimicrob. Agents Chemother. 2016, 60, 862–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rungelrath, V.; DeLeo, F.R. Staphylococcus aureus, Antibiotic Resistance, and the Interaction with Human Neutrophils. Antioxid. Redox Signal. 2021, 34, 452–470. [Google Scholar] [CrossRef]
- Straniero, V.; Suigo, L.; Casiraghi, A.; Sebastián-Pérez, V.; Hrast, M.; Zanotto, C.; Zdovc, I.; De Giuli Morghen, C.; Radaelli, A.; Valoti, E. Benzamide Derivatives Targeting the Cell Division Protein FtsZ: Modifications of the Linker and the Benzodioxane Scaffold and Their Effects on Antimicrobial Activity. Antibiotics 2020, 9, 160. [Google Scholar] [CrossRef] [Green Version]
- Holmes, N.E.; Johnson, P.D.R.; Howden, B.P. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J. Clin. Microbiol. 2012, 50, 2548–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, T.J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 2017, 41, 430–449. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, L.; Fu, J.; Yan, M.; Chen, D.; Zhang, L. Microbial pathogenicity and virulence mediated by integrons on Gram-positive microorganisms. Microb. Pathog. 2017, 111, 481–486. [Google Scholar] [CrossRef]
- Solomon, S.L.; Oliver, K.B. Antibiotic Resistance Threats in the United States: Stepping Back from the Brink. AFP 2014, 89, 938–941. [Google Scholar]
- McGuinness, W.A.; Malachowa, N.; DeLeo, F.R. Vancomycin Resistance in Staphylococcus aureus. Yale J. Biol. Med. 2017, 90, 269–281. [Google Scholar]
- Straniero, V.; Sebastián Pérez, V.; Hrast, M.; Zanotto, C.; Casiraghi, A.; Suigo, L.; Zdovc, I.; Radaelli, A.; De Giuli Morghen, C.; Valoti, E. Benzodioxane-benzamides as antibacterial agents: Computational and SAR studies to evaluate the influence of the 7-substitution in FtsZ interaction. ChemMedChem 2020, 2, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, R.P. The antibiotic pipeline—Challenges, costs, and values. N. Engl. J. Med. 2004, 351, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.; Petfalski, E.; Shevchenko, A.; Mann, M.; Tollervey, D. The Exosome: A Conserved Eukaryotic RNA Processing Complex Containing Multiple 3’→5’ Exoribonucleases. Cell 1997, 91, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Tejada-Arranz, A.; de Crécy-Lagard, V.; de Reuse, H. Bacterial RNA Degradosomes: Molecular Machines under Tight Control. Trends Biochem. Sci. 2020, 45, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, C.; Wu, M.; Tian, T.; Cheng, T.; Zhang, X.; Zang, J. Enolase binds to RnpA in competition with PNPase in Staphylococcus aureus. FEBS Lett. 2017, 591, 3523–3535. [Google Scholar] [CrossRef] [Green Version]
- Roux, C.M.; DeMuth, J.P.; Dunman, P.M. Characterization of components of the Staphylococcus aureus mRNA degradosome holoenzyme-like complex. J. Bacteriol. 2011, 193, 5520–5526. [Google Scholar] [CrossRef] [Green Version]
- Olson, P.D.; Kuechenmeister, L.J.; Anderson, K.L.; Daily, S.; Beenken, K.E.; Roux, C.M.; Reniere, M.L.; Lewis, T.L.; Weiss, W.J.; Pulse, M.; et al. Small Molecule Inhibitors of Staphylococcus aureus RnpA Alter Cellular mRNA Turnover, Exhibit Antimicrobial Activity, and Attenuate Pathogenesis. PLoS Pathog. 2011, 7, e1001287. [Google Scholar] [CrossRef] [Green Version]
- Carpousis, A.J. The RNA degradosome of Escherichia coli: An mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 2007, 61, 71–87. [Google Scholar] [CrossRef] [Green Version]
- Eidem, T.M.; Lounsbury, N.; Emery, J.F.; Bulger, J.; Smith, A.; Abou-Gharbia, M.; Childers, W.; Dunman, P.M. Small-molecule inhibitors of Staphylococcus aureus RnpA-mediated RNA turnover and tRNA processing. Antimicrob. Agents Chemother. 2015, 59, 2016–2028. [Google Scholar] [CrossRef] [Green Version]
- Lounsbury, N.; Eidem, T.; Colquhoun, J.; Mateo, G.; Abou-Gharbia, M.; Dunman, P.M.; Childers, W.E. Novel inhibitors of Staphylococcus aureus RnpA that synergize with mupirocin. Bioorg. Med. Chem. Lett. 2018, 28, 1127–1131. [Google Scholar] [CrossRef]
- Colquhoun, J.M.; Ha, L.; Beckley, A.; Meyers, B.; Flaherty, D.P.; Dunman, P.M. Identification of Small Molecule Inhibitors of Staphylococcus aureus RnpA. Antibiotics 2019, 8, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naclerio, G.A.; Abutaleb, N.S.; Li, D.; Seleem, M.N.; Sintim, H.O. Ultrapotent Inhibitor of Clostridioides difficile Growth, Which Suppresses Recurrence In Vivo. J. Med. Chem. 2020, 63, 11934–11944. [Google Scholar] [CrossRef] [PubMed]
- Ha, L.; Colquhoun, J.; Noinaj, N.; Das, C.; Dunman, P.M.; Flaherty, D.P. Crystal structure of the ribonuclease-P-protein subunit from Staphylococcus aureus. Acta Crystallogr. F Struct. Biol. Commun. 2018, 74, 632–637. [Google Scholar] [CrossRef] [PubMed]
- DeBons, F.E.; Loudon, G.M. Protected diaminomethane. J. Org. Chem. 1980, 45, 1703–1704. [Google Scholar] [CrossRef]
- Radoux, C.J.; Olsson, T.S.G.; Pitt, W.R.; Groom, C.R.; Blundell, T.L. Identifying Interactions that Determine Fragment Binding at Protein Hotspots. J. Med. Chem. 2016, 59, 4314–4325. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | MSSA (ATCC 29213) | MRSA (ATCC 43300) |
|---|---|---|
| MIC (μM) | MIC (μM) | |
| 1 | >500 | >500 |
| 2 | >500 | >500 |
| 3 | >500 | >500 |
| 4 | 311 | 311 |
| 5 | >500 | >500 |
| 6 | 21.1 | 21.1 |
| 7 | 24.7 | 24.7 |
| 8 | 22.2 | 22.2 |
| 9 | >500 | 18.9 |
| 10 | 21.1 | 21.1 |
| 11 | >500 | >500 |
| 12 | >500 | >500 |
| 13 | >500 | >500 |
| 14 | >500 | >500 |
| Compound Name | Degradation IC50 * | Processing IC50 ** |
|---|---|---|
| RNPA2000 | 275 | 140 |
| 1 | 72.5 | 36 |
| 2 | 233 | 37 |
| 3 | 324 | >500 |
| 4 | 66 | 50 |
| 5 | >500 | 75 |
| 6 | 53 | 59 |
| 7 | 77 | 28 |
| 8 | 49 | 76 |
| 9 | - | - |
| 10 | 188 | 33 |
| 11 | 31 | 153 |
| 12 | 165 | 423 |
| 13 | 198 | >500 |
| 14 | 174 | 25 |
| Method Name | Mobile Phase | Flow Rate (mL/min) |
|---|---|---|
| A | H2O (1‰ TFA)/ACN (1‰ TFA) 55/45 | 1 |
| B | H2O (1‰ TFA)/ACN (1‰ TFA) 55/45 | 1.5 |
| C | H2O (1‰ TFA)/ACN (1‰ TFA) 1/1 | 1.5 |
| D | H2O (1‰ TFA)/ACN (1‰ TFA) 40/60 | 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suigo, L.; Chojnacki, M.; Zanotto, C.; Sebastián-Pérez, V.; Morghen, C.D.G.; Casiraghi, A.; Dunman, P.M.; Valoti, E.; Straniero, V. Staphylococcus aureus RnpA Inhibitors: Computational-Guided Design, Synthesis and Initial Biological Evaluation. Antibiotics 2021, 10, 438. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10040438
Suigo L, Chojnacki M, Zanotto C, Sebastián-Pérez V, Morghen CDG, Casiraghi A, Dunman PM, Valoti E, Straniero V. Staphylococcus aureus RnpA Inhibitors: Computational-Guided Design, Synthesis and Initial Biological Evaluation. Antibiotics. 2021; 10(4):438. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10040438
Chicago/Turabian StyleSuigo, Lorenzo, Michaelle Chojnacki, Carlo Zanotto, Victor Sebastián-Pérez, Carlo De Giuli Morghen, Andrea Casiraghi, Paul M. Dunman, Ermanno Valoti, and Valentina Straniero. 2021. "Staphylococcus aureus RnpA Inhibitors: Computational-Guided Design, Synthesis and Initial Biological Evaluation" Antibiotics 10, no. 4: 438. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10040438