Structural Elucidation of Antibiotic TKR2999, an Antifungal Lipodepsipeptide Isolated from the Fungus Foliophoma fallens


 , ,
, ,  , , , and
, , , and 
Abstract
:
1. Introduction
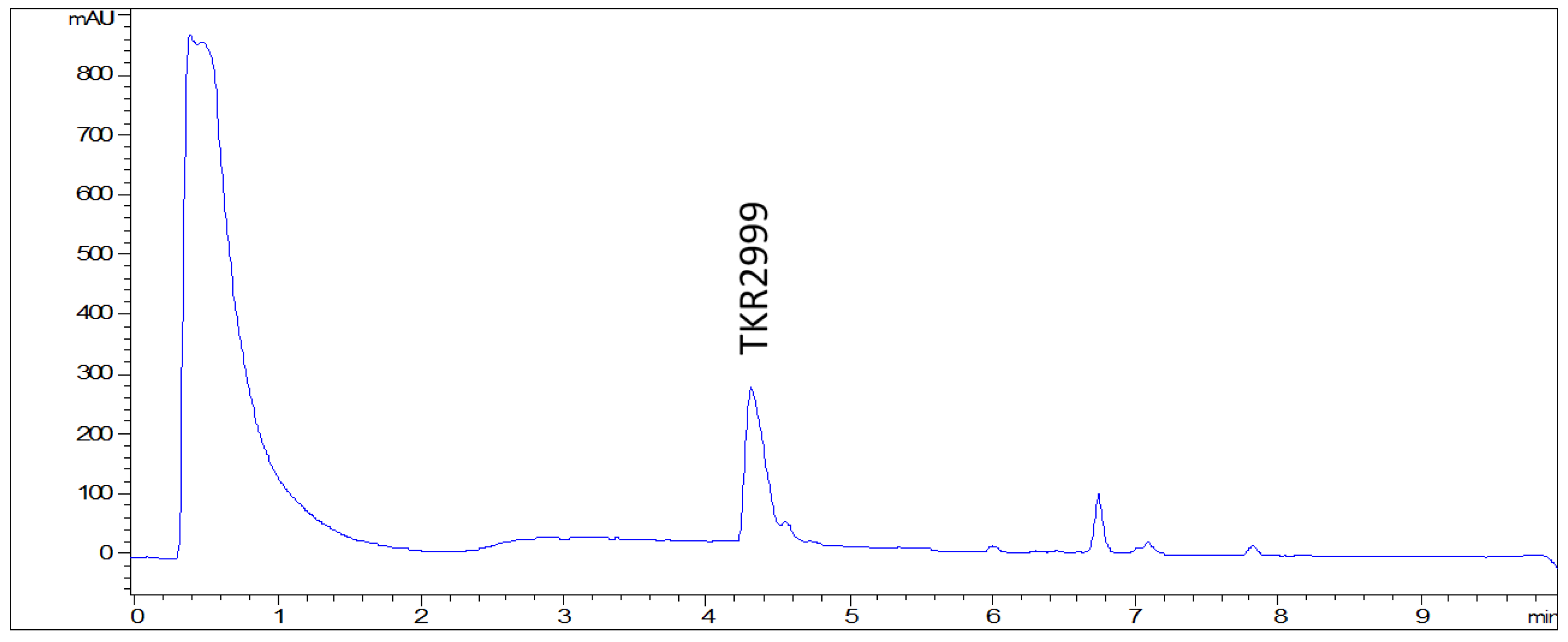
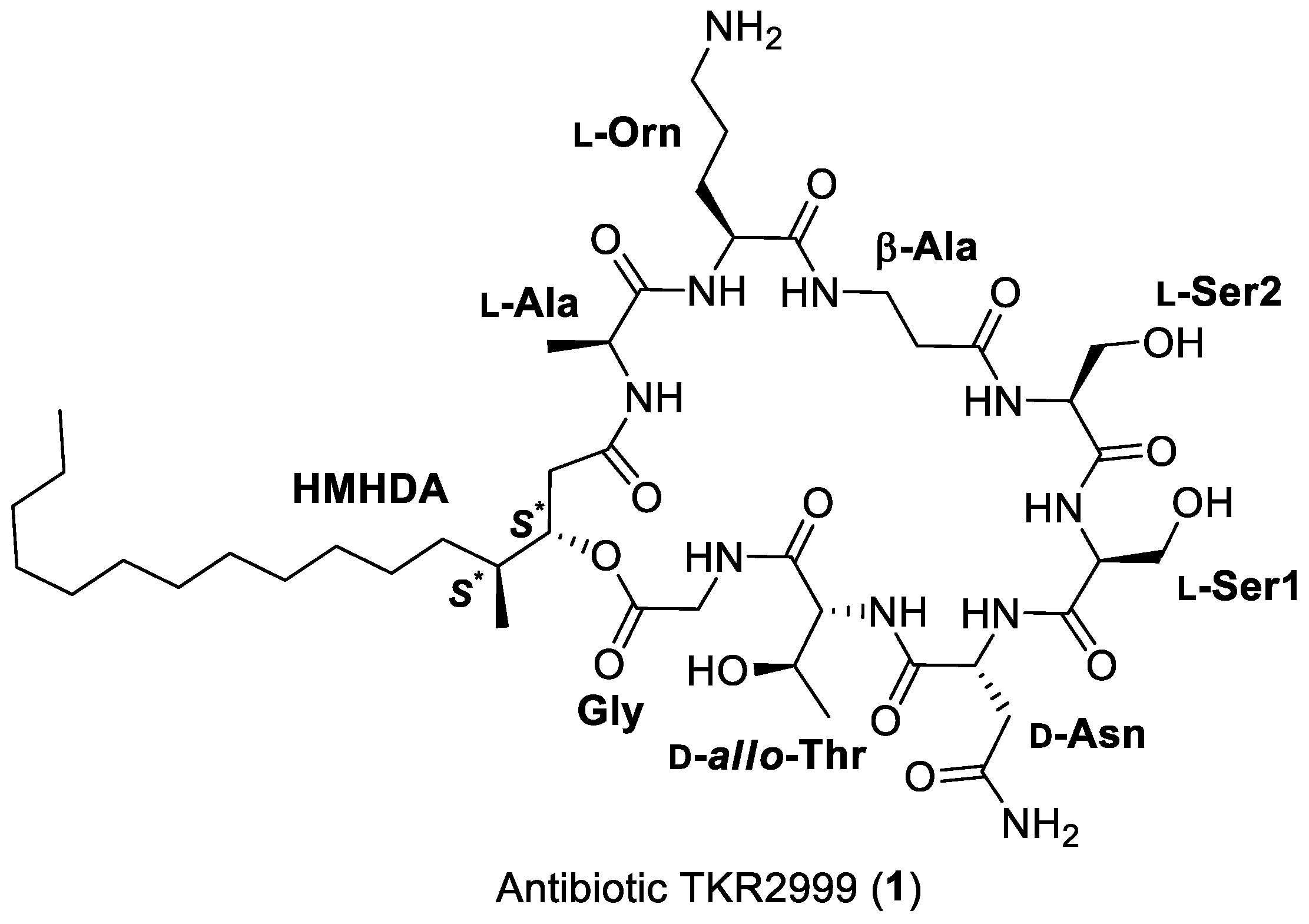
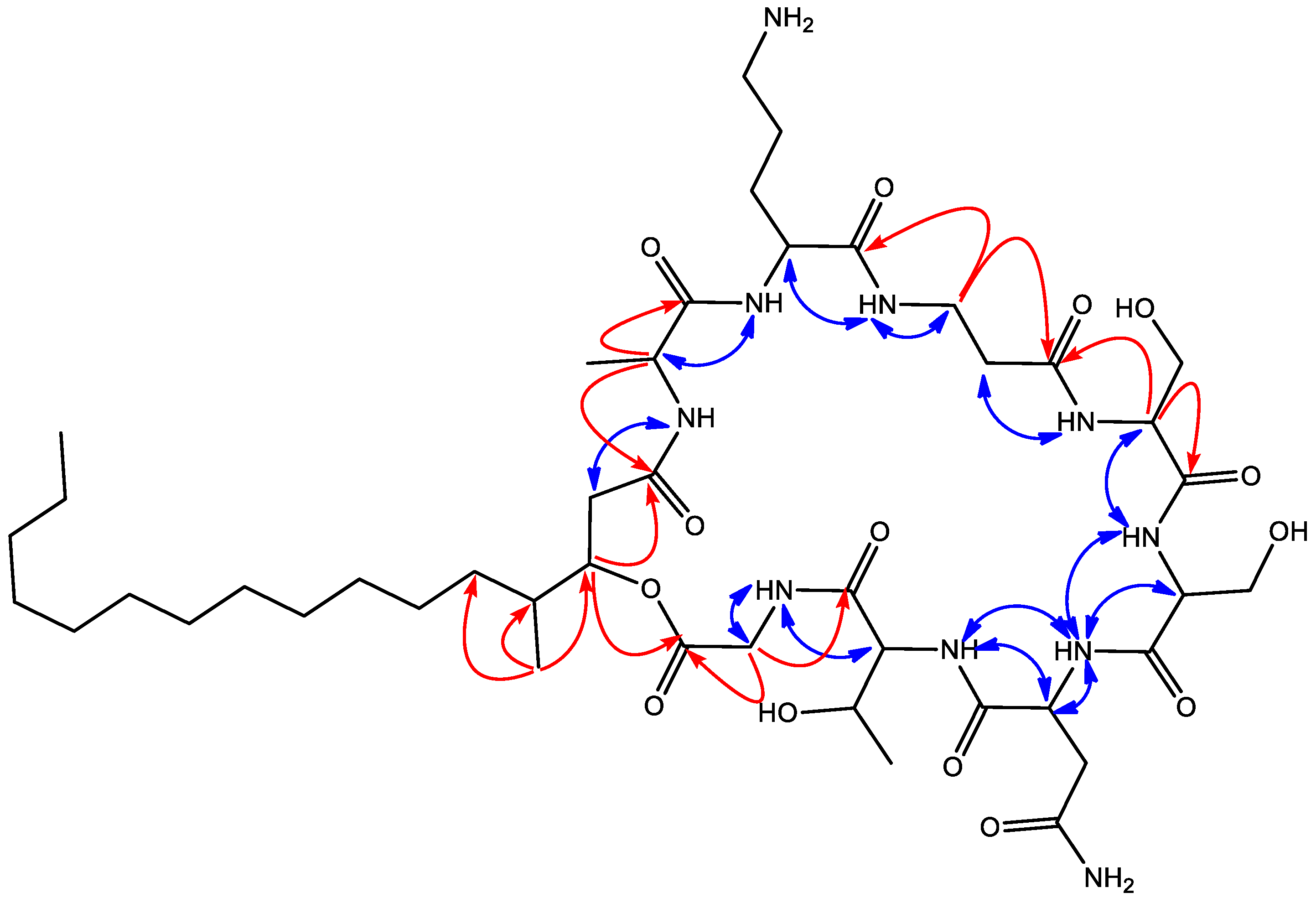
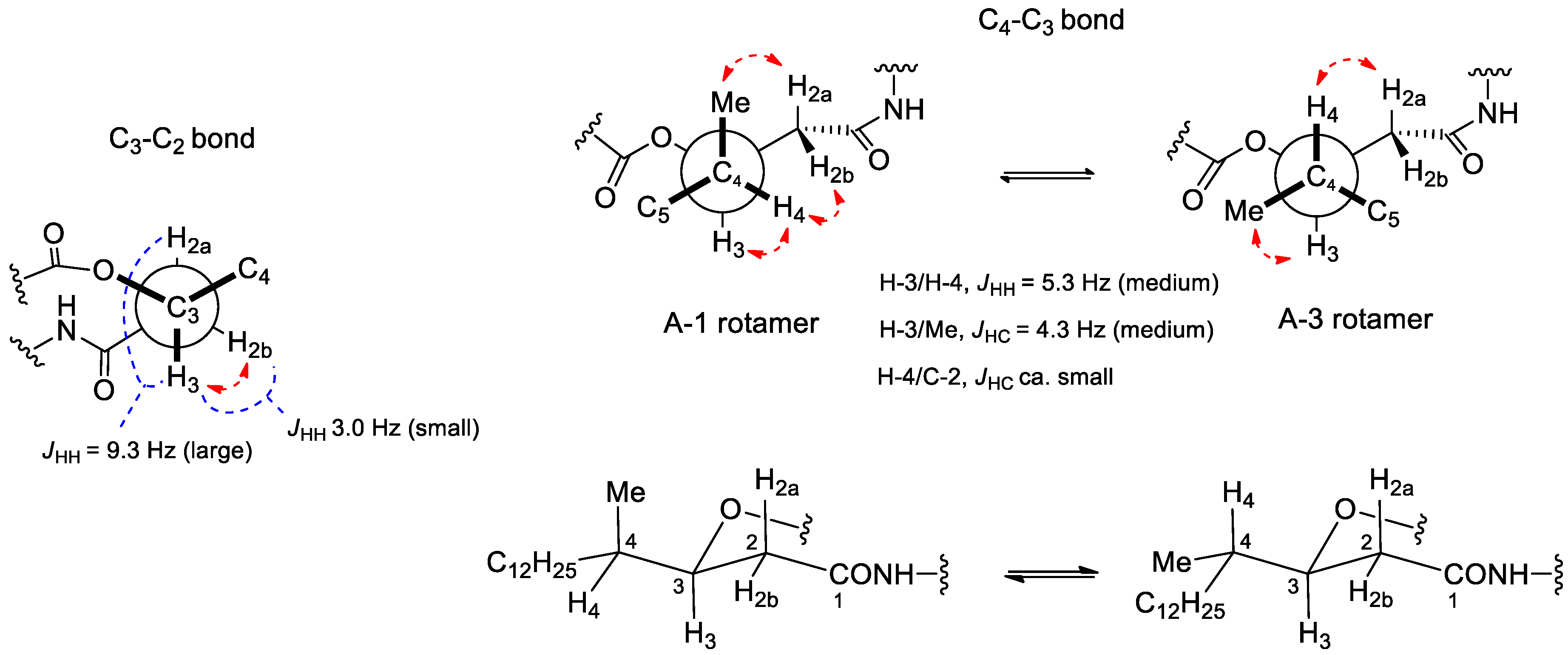
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Producing Fungus and Its Characterization
3.3. Fermentation
3.4. Extraction and Isolation
3.5. Marfey’s Analysis of Compound 1
3.6. Biological Activity
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lackner, M.; Lass-Florl, C. Up-date on diagnostic strategies of invasive aspergillosis. Curr. Pharm. Des. 2013, 19, 3595–3614. [Google Scholar] [CrossRef] [PubMed]
- Cowen, L.E.; Steinbach, W.J. Stress, drugs, and evolution: The role of cellular signaling in fungal drug resistance. Eukaryot. Cell 2008, 7, 747–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkovec, J.M.; Hughes, D.L.; Masurekar, P.S.; Sable, C.A.; Schwartz, R.E.; Singh, S.B. Discovery and development of first in class antifungal caspofungin (CANCIDAS®)-A case study. Nat. Prod. Rep. 2014, 31, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.A.; Slavin, M.; Sorrell, T. Echinocandin Antifungal Drugs in Fungal Infections. Drugs 2011, 71, 11–41. [Google Scholar] [CrossRef]
- Crespo, G.; González-Menéndez, V.; de la Cruz, M.; Martín, J.; Cautain, B.; Sánchez, P.; Pérez-Victoria, I.; Vicente, F.; Genilloud, O.; Reyes, F. Antifungal long-chain alkenyl sulphates isolated from culture broths of the fungus Chaetopsina sp. Planta Med. 2017, 234, 545–550. [Google Scholar] [CrossRef]
- Takesako, K.; Awazu, N.; Ueno, M.; Onishi, Y.; Kato, I. Antibiotic TKR2999, process for the preparation thereof and microbe. Japanese Patent WO99/32498 A1, 1 July 1999. [Google Scholar]
- Singh, S.B.; Ondeyka, J.; Harris, G.; Herath, K.; Zink, D.; Vicente, F.; Bills, G.; Collado, J.; Platas, G.; González del Val, A.; et al. Isolation, structure, and biological activity of phaeofungin, a cyclic lipodepsipeptide from a Phaeosphaeria sp. Using the genome-wide Candida albicans fitness test. J. Nat. Prod. 2013, 76, 334–345. [Google Scholar] [CrossRef]
- Herath, K.; Harris, G.; Jayasuriya, H.; Zink, D.; Smith, S.; Vicente, F.; Bills, G.; Collado, J.; González, A.; Jiang, B.; et al. Isolation, structure and biological activity of phomafungin, a cyclic lipodepsipeptide from a widespread tropical Phoma sp. Bioorg. Med. Chem. 2009, 17, 1361–1369. [Google Scholar] [CrossRef]
- Crous, P.W.; Groenewald, J.Z. The Genera of Fungi–G 4: Camarosporium and Dothiora. IMA Fungus 2017, 8, 131–152. [Google Scholar] [CrossRef] [Green Version]
- Hyde, K.D.; Dong, Y.; Phookamsak, R.; Jeewon, R.; Bhat, D.J.; Jones, E.B.G.; Liu, N.-G.; Abeywickrama, P.D.; Mapook, A.; Wei, D.; et al. Fungal diversity notes 1151–1276: Taxonomic and phylogenetic contributions on genera and species of fungal taxa. Fungal Divers. 2020, 100, 5–277. [Google Scholar] [CrossRef] [Green Version]
- Marfey, P. Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984, 49, 591–596. [Google Scholar] [CrossRef] [Green Version]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, G.R.; Dale, J.A.; Mosher, H.S. Correlation of configuration and fluorine-19 chemical shifts of -methoxy- -trifluoromethylphenyl acetate derivatives. J. Org. Chem. 1973, 38, 2143–2147. [Google Scholar] [CrossRef]
- Meissner, A.; Sørensen, O.W. Measurement of J(H,H) and long-range J(X,H) coupling constants in small molecules. Broadband XLOC and J-HMBC. Magn. Reson. Chem. 2001, 39, 49–52. [Google Scholar] [CrossRef]
- Haasnoot, C.A.G.; de Leeuw, F.A.A.M.; Altona, C. The relationship between proton-proton NMR coupling constants and substituent electronegativities—I: An empirical generalization of the karplus equation. Tetrahedron 1980, 36, 2783–2792. [Google Scholar] [CrossRef]
- Aydin, R.; Günther, H. 13C, 1H spin–spin coupling. X—Norbornane: A reinvestigation of the karplus curve for 3J(13C, 1H). Magn. Reson. Chem. 1990, 28, 448–457. [Google Scholar] [CrossRef]
- Ding, L.-J.; Yuan, W.; Liao, X.-J.; Han, B.-N.; Wang, S.-P.; Li, Z.-Y.; Xu, S.-H.; Zhang, W.; Lin, H.-W. Oryzamides A–E, cyclodepsipeptides from the sponge-derived fungus Nigrospora oryzae PF18. J. Nat. Prod. 2016, 79, 2045–2052. [Google Scholar] [CrossRef]
- Elbanna, A.H.; Khalil, Z.G.; Bernhardt, P.V.; Capon, R.J. Scopularides revisited: Molecular networking guided exploration of lipodepsipeptides in australian marine fish gastrointestinal tract-derived fungi. Mar. Drugs 2019, 17, 475. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.-M.; Li, Y.-Y.; Shi, Y.-W.; Fang, Y.-W.; Chao, R.; Gu, Y.-C.; Wang, C.-Y.; Shao, C.-L. Integrating molecular networking and 1H NMR to target the isolation of chrysogeamides from a library of marine-derived Penicillium fungi. J. Org. Chem. 2019, 84, 1228–1237. [Google Scholar] [CrossRef]
- Vicente, F.; Reyes, F.; Genilloud, O. Fungal secondary metabolites as source of antifungal compounds. In Fungi Applications and Management Strategies; Chapter 5; Deshmukh, S.K., Misra, J.K., Tewari, J.P., Papp, T., Eds.; CRC Press, Taylor & Francis Group: Boca Ratón, FL, USA, 2016; pp. 80–116. [Google Scholar]
- Collado, J.; Platas, G.; Paulus, B.; Bills, G.F. High-throughput culturing of fungi from plant litter by a dilution-to-extinction technique. FEMS Microbiol. Ecol. 2007, 60, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Menendez, V.; Martin, J.; Siles, J.A.; Gonzalez-Tejero, M.R.; Reyes, F.; Platas, G.; Tormo, J.R.; Genilloud, O. Biodiversity and chemotaxonomy of Preussia isolates from the Iberian Peninsula. Mycol. Prog. 2017, 16, 713–728. [Google Scholar] [CrossRef]
- Biological Resource Center, National Institute of Technology and Evaluation. Available online: https://www.nite.go.jp/en/nbrc/index.html (accessed on 25 May 2020).
- Pairwise Sequence Alignment, CBS-KNAW Collections. Available online: http://www.wi.knaw.nl/Collections/BioloMICSSequences.aspx?expandparm=f&file=FUNGI&file=FUNCBS&file=YEASTseq&file=MOLDseq&file=UNITE&file=Yeast&file=Fusarium&file=Dermato&file=indoor&file=Morch&file=ITSSeq&file=Bipolaris&file=CGattii&file=CNeof&file=PJ&file=Scedo&file=FunBOLD&file=PYCC&file=EFSeq&file=Mycokey&file=PasteurMOULD&file=PasteurYEAST (accessed on 25 May 2020).
- Monteiro, M.C.; de la Cruz, M.; Cantizani, J.; Moreno, C.; Tormo, J.R.; Mellado, E.; De Lucas, J.R.; Asensio, F.; Valiante, V.; Brakhage, A.A.; et al. A new approach to drug discovery: High-throughput screening of microbial natural extracts against Aspergillus fumigatus using resazurin. J. Biomol. Screen. 2012, 17, 542–549. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC | δH, Mult (J in Hz) | Position | δC | δH, Mult (J in Hz) |
|---|---|---|---|---|---|
| Gly | Orn | ||||
| 1 | 169.0 | 1 | 170.9 | ||
| 2 | 40.9 | 3.98 dd (17.4, 4.6) 3.72 dd (17.4, 6.3) | 2 | 52.0 | 4.20 m |
| NH | 8.16 t (5.3) | 3 | 28.5 | 1.77 m 1.52 m | |
| Thr | 4 | 23.6 | 1.52 m | ||
| 1 | 170.2 | 5 | 38.6 | 2.75 m | |
| 2 | 58.3 | 4.24 m | NH | 7.99 m | |
| 3 | 66.8 | 3.95 m | NH2 | 7.63 m | |
| 4 | 19.9 | 1.07 d (6.2) | Ala | ||
| NH | 7.67 d (8.7) | 1 | 172.5 | ||
| 3-OH | 4.87 d (4.3) | 2 | 49.0 | 4.17 m | |
| Asn | 3 | 17.9 | 1.23 m | ||
| 1 | 170.6 | NH | 7.99 m | ||
| 2 | 50.0 | 4.57 m | HMHDA | ||
| 3 | 36.9 | 2.65 dd (15.5, 5.7) 2.50 m | 1 | 169.8 | |
| 4 | 172.1 | 2 | 36.7 | 2.39 dd (14.4, 9.3) a 2.31 dd (14.4, 2.7) a | |
| NH | 8.32 d (8.0) | 3 | 75.2 | 5.04 ddd (8.6, 5.3, 3.2) a | |
| NH2 | 7.43 s 7.01 s | 4 | 35.8 | 1.68 m | |
| Ser1 | 54 | 31.6 | 1.31 m 1.04 m | ||
| 1 | 170.1 | 6 | 26.5 | 1.31 m 1.17 m | |
| 2 | 56.0 | 4.23 m | 7 | 29.2 | 1.24 m |
| 3 | 61.2 | 3.63 m 3.59 m | 8 | 29.1 | 1.24 m |
| NH | 7.99 m | 9 | 29.1 | 1.24 m | |
| 3-OH | 4.94 t (4.9) | 10 | 29.1 | 1.24 m | |
| Ser2 | 11 | 29.1 | 1.24 m | ||
| 1 | 170.7 | 12 | 29.1 | 1.24 m | |
| 2 | 55.3 | 4.30 m | 13 | 28.7 | 1.24 m |
| 3 | 61.5 | 3.63 m 3.54 m | 14 | 31.3 | 1.23 m |
| NH | 8.03 d (7.6) | 15 | 22.1 | 1.26 m | |
| 3-OH | 5.04 m | 16 | 14.0 | 0.85 t (6.8) | |
| β-Ala | 4-Me | 14.7 | 0.82 d (6.6) | ||
| 1 | 171.3 | ||||
| 2 | 35.1 | 2.36 m | |||
| 3 | 35.5 | 3.34 m 3.29 m | |||
| NH | 7.76 t (5.3) |
| Fungal Strain/Cell Line | |||||||
|---|---|---|---|---|---|---|---|
| Compound | C. glabrata | C. krusei | C. parapsilosis | C. tropicalis | C. albicans | A. fumigatus | THLE-2 |
| MIC (µg/mL) | IC50 (µg/mL) | ||||||
| 1 | 1–0.5 | 2–1 | 2–1 | 1–0.5 | 1 | 1–0.5 | 5.38 ± 0.91 |
| Amphotericin B | 2 | 4 | 4 | 4 | 4 | 4 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crespo, G.; Pérez-Victoria, I.; Ortiz-López, F.J.; González-Menéndez, V.; de la Cruz, M.; Cautain, B.; Sánchez, P.; Vicente, F.; Genilloud, O.; Reyes, F. Structural Elucidation of Antibiotic TKR2999, an Antifungal Lipodepsipeptide Isolated from the Fungus Foliophoma fallens. Antibiotics 2020, 9, 278. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060278
Crespo G, Pérez-Victoria I, Ortiz-López FJ, González-Menéndez V, de la Cruz M, Cautain B, Sánchez P, Vicente F, Genilloud O, Reyes F. Structural Elucidation of Antibiotic TKR2999, an Antifungal Lipodepsipeptide Isolated from the Fungus Foliophoma fallens. Antibiotics. 2020; 9(6):278. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060278
Chicago/Turabian StyleCrespo, Gloria, Ignacio Pérez-Victoria, Francisco Javier Ortiz-López, Víctor González-Menéndez, Mercedes de la Cruz, Bastien Cautain, Pilar Sánchez, Francisca Vicente, Olga Genilloud, and Fernando Reyes. 2020. "Structural Elucidation of Antibiotic TKR2999, an Antifungal Lipodepsipeptide Isolated from the Fungus Foliophoma fallens" Antibiotics 9, no. 6: 278. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060278