
Effect of Alkali Treatment on the Properties of Acacia Caesia Bark Fibres

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Treatament
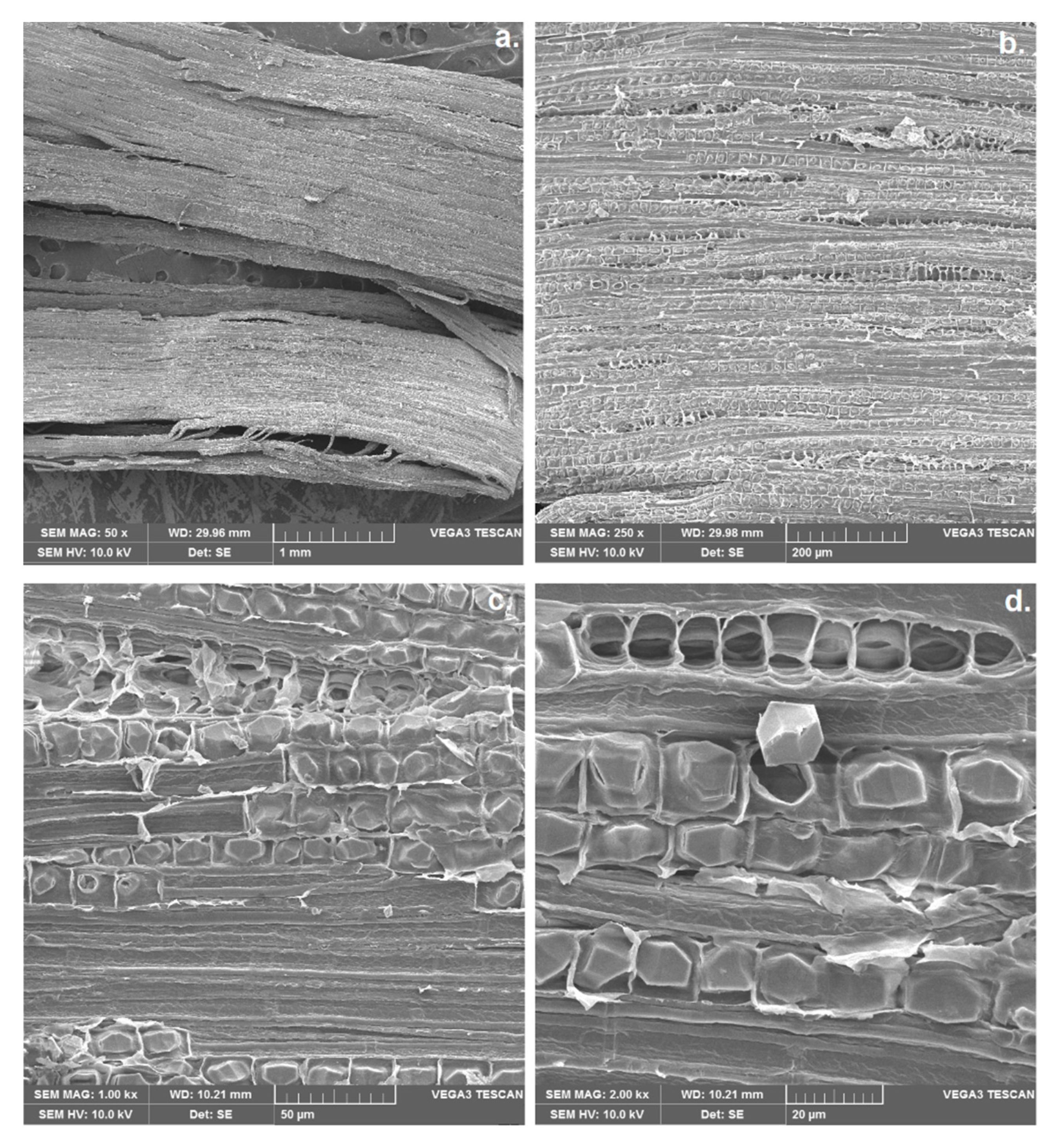
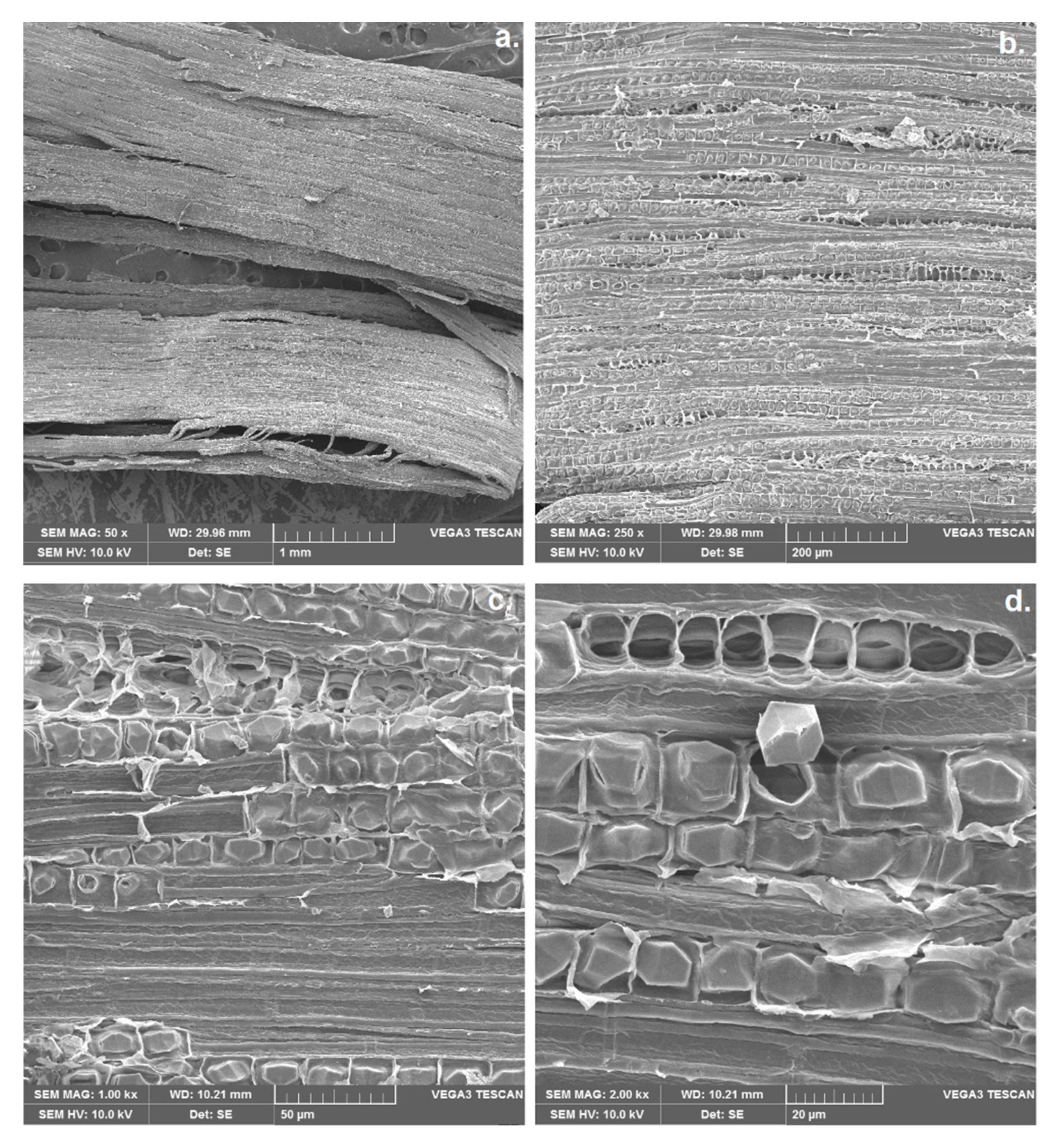
2.2. Scanning Electron Microscopy (SEM)
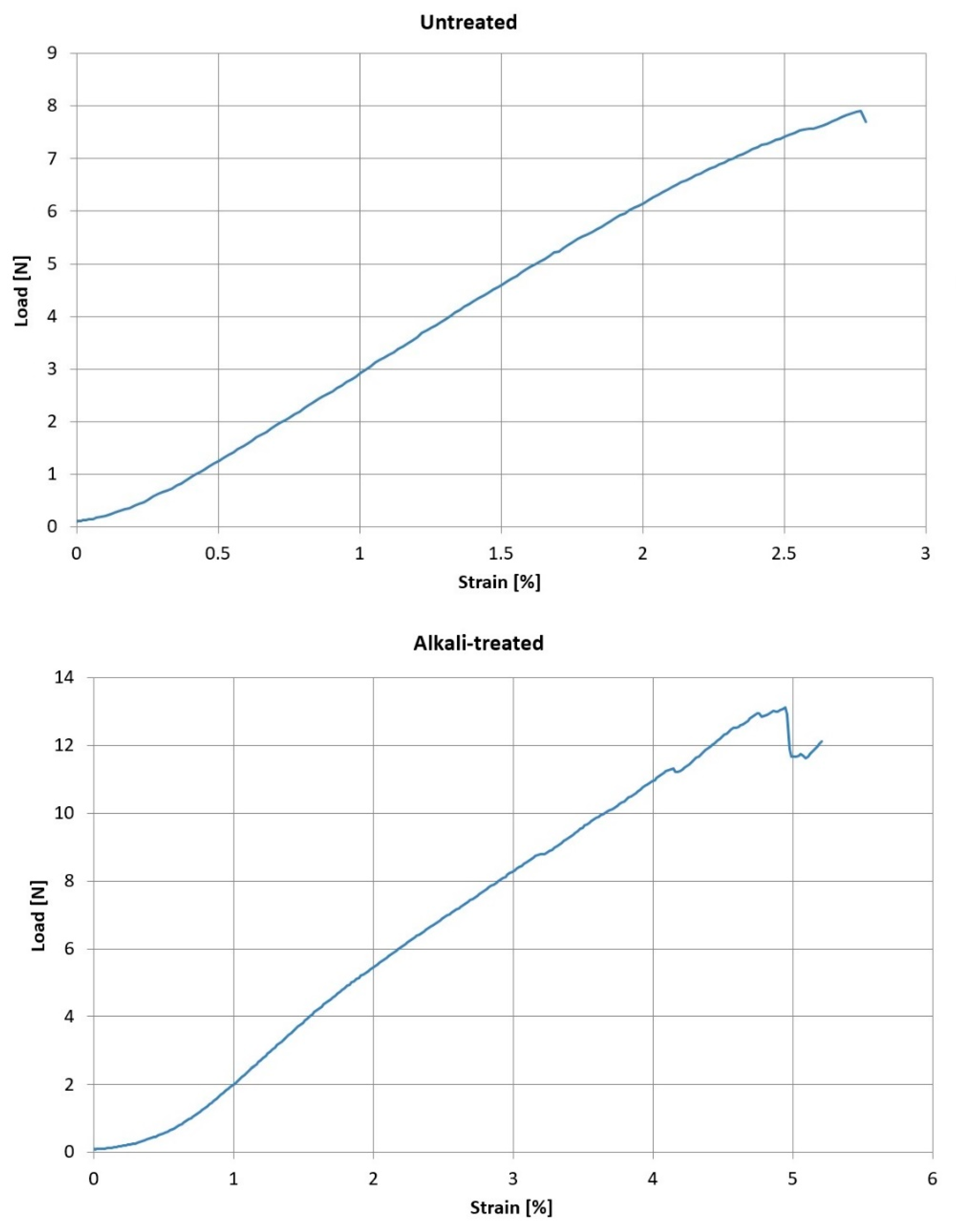
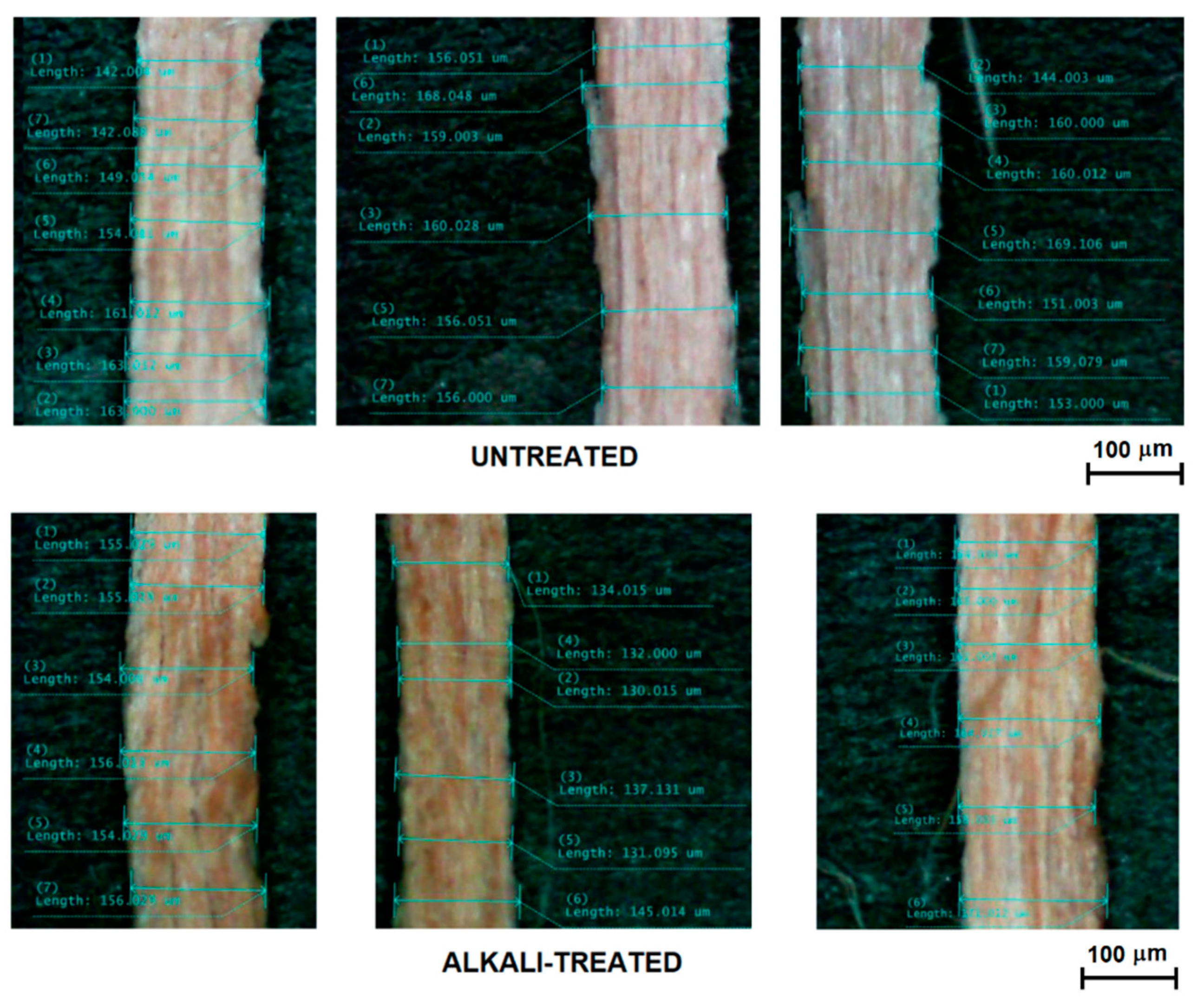
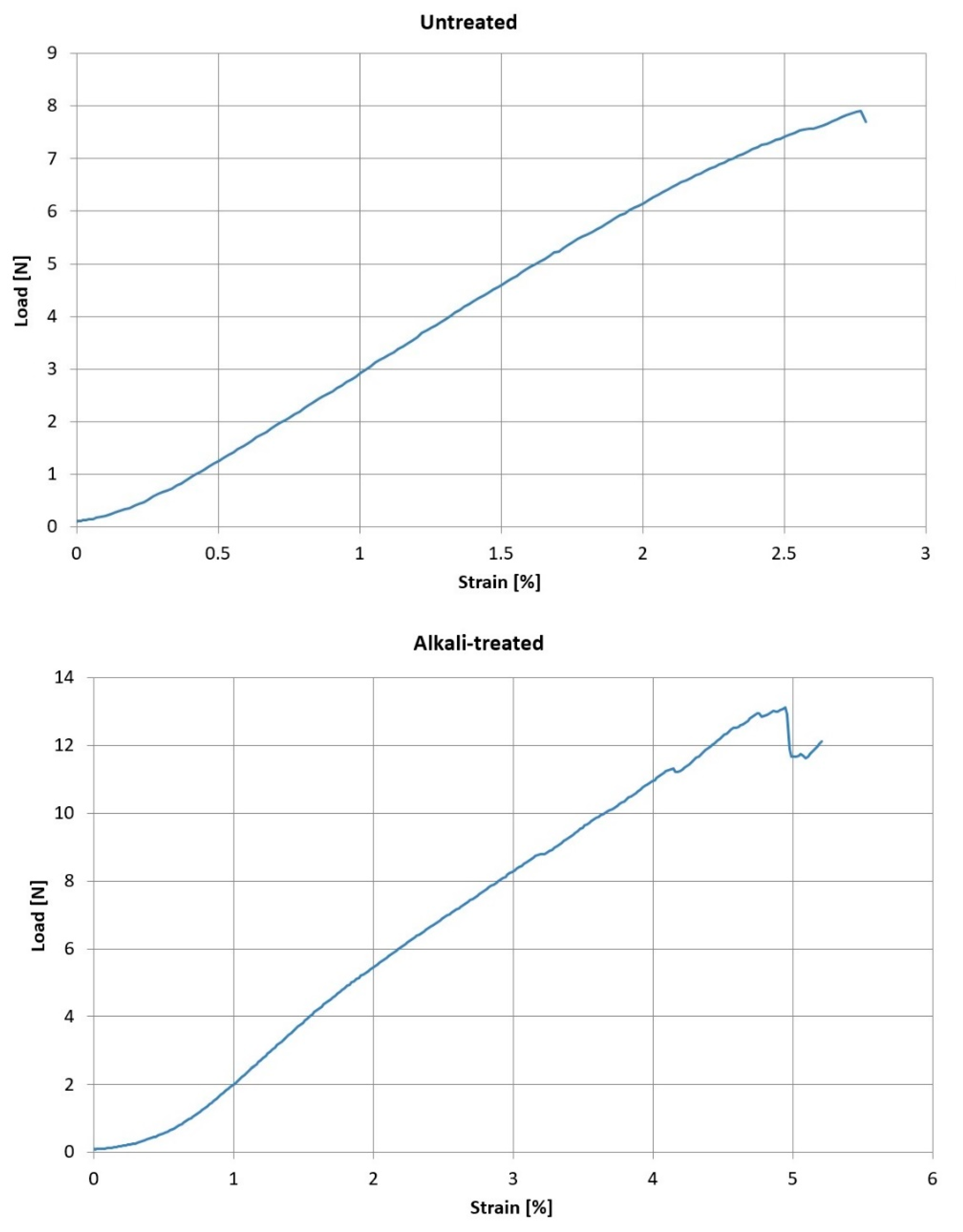
2.3. Single Fibre Tensile Testing (SFTT)
2.4. Chemical and Physical Characterization
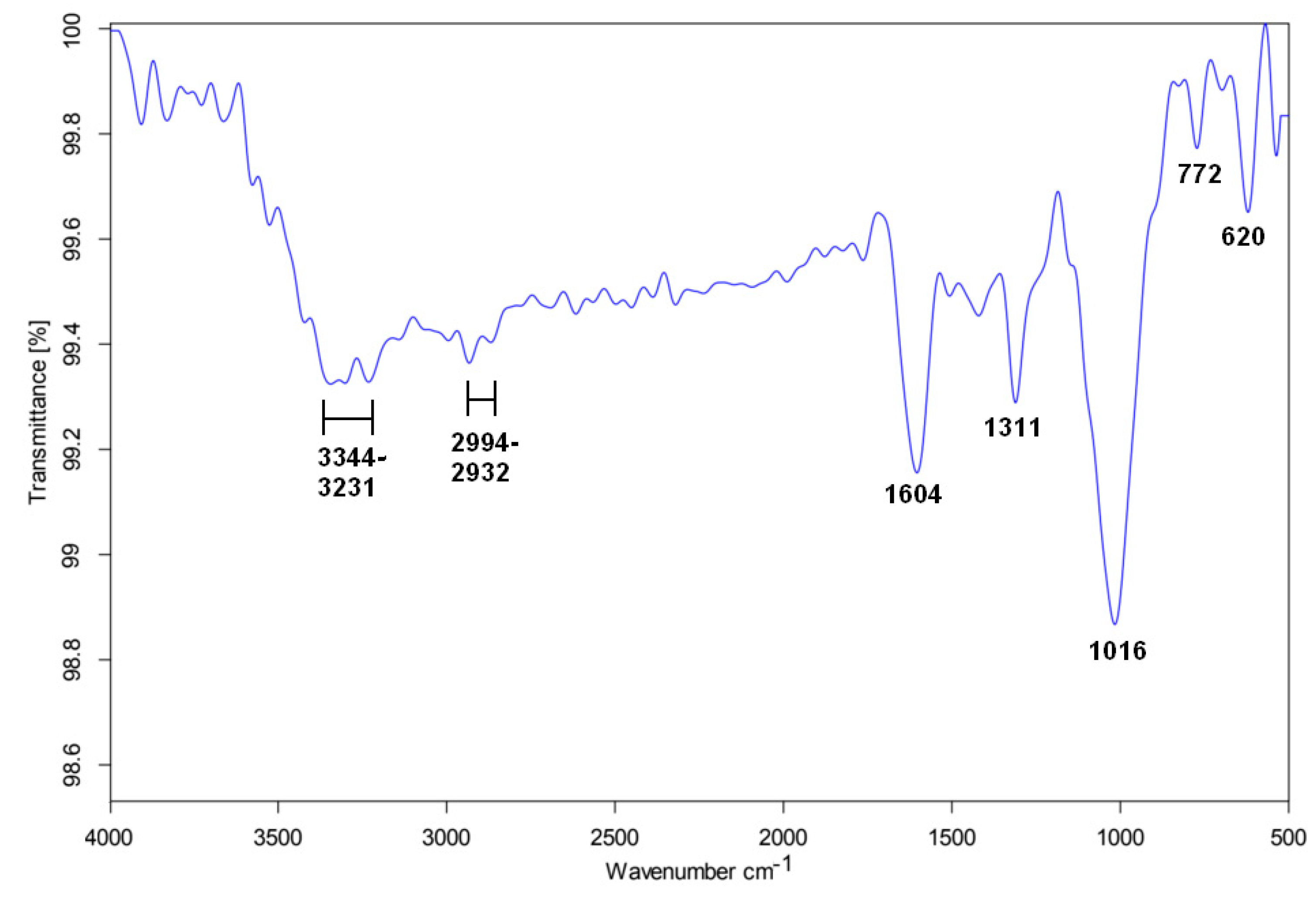
2.5. Fourier Transform Infrared (FTIR) Spectroscopy
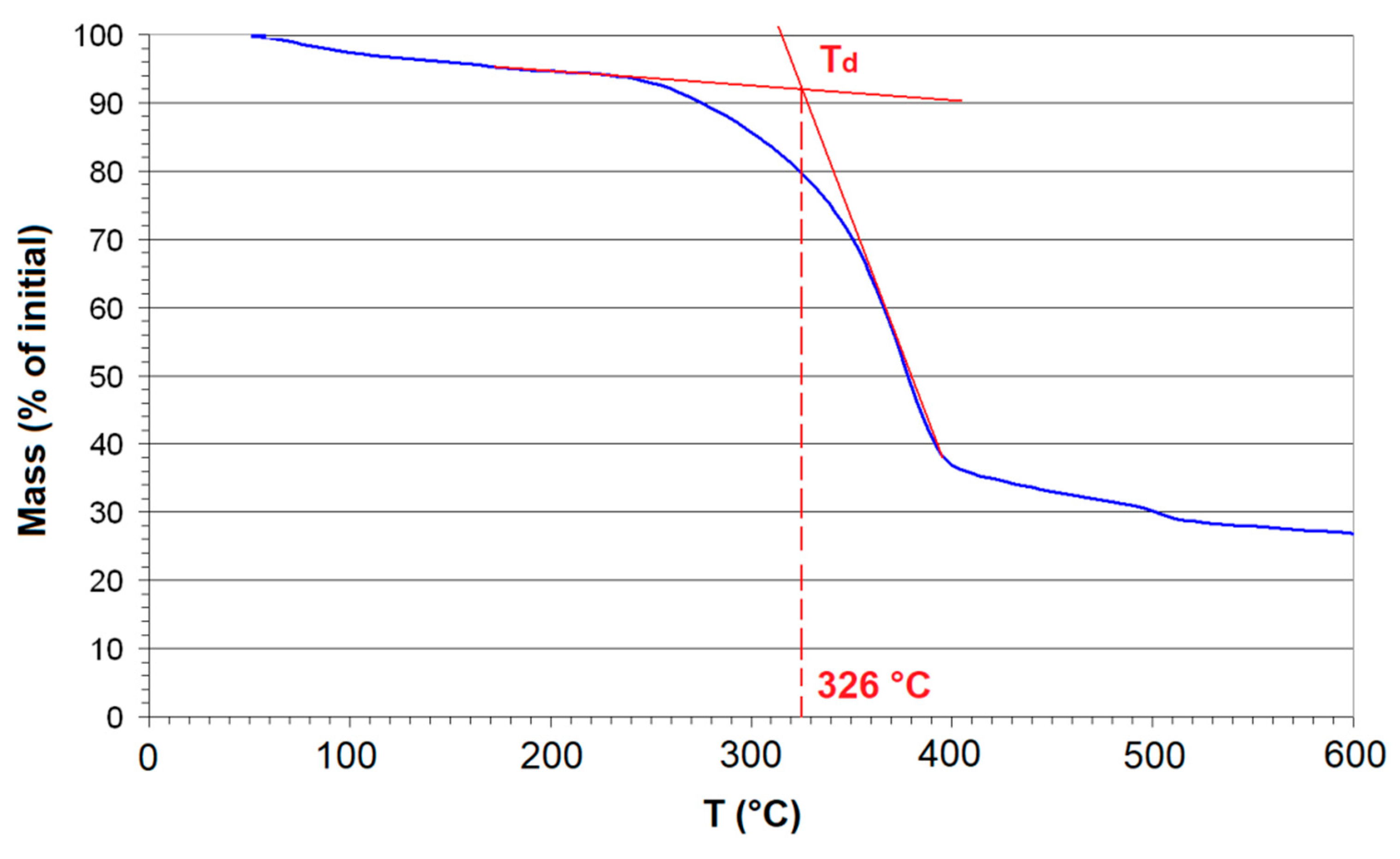
2.6. Thermogravimetric Analysis (TGA)
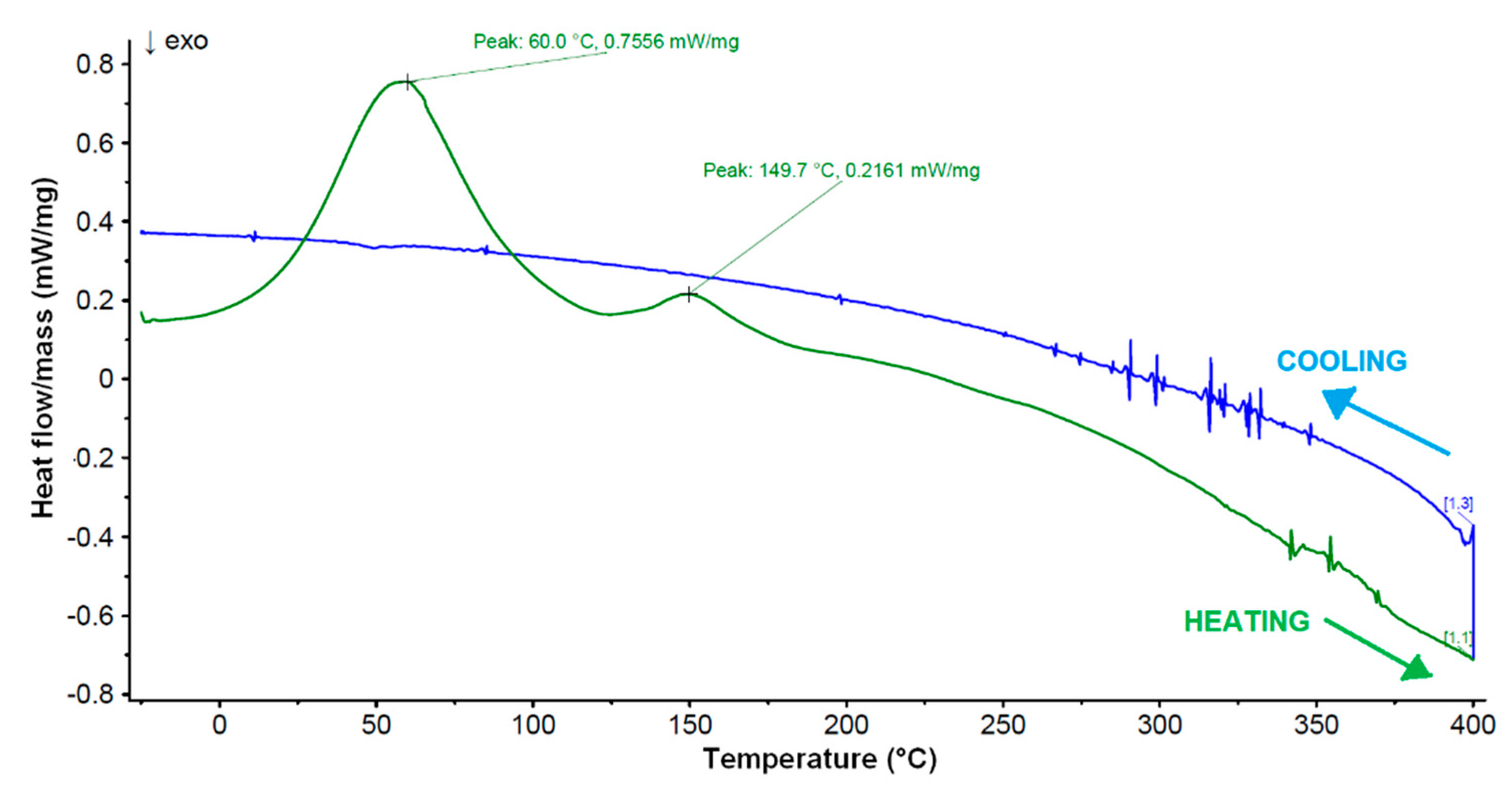
2.7. Differential Scanning Calorimetry (DSC) Analysis
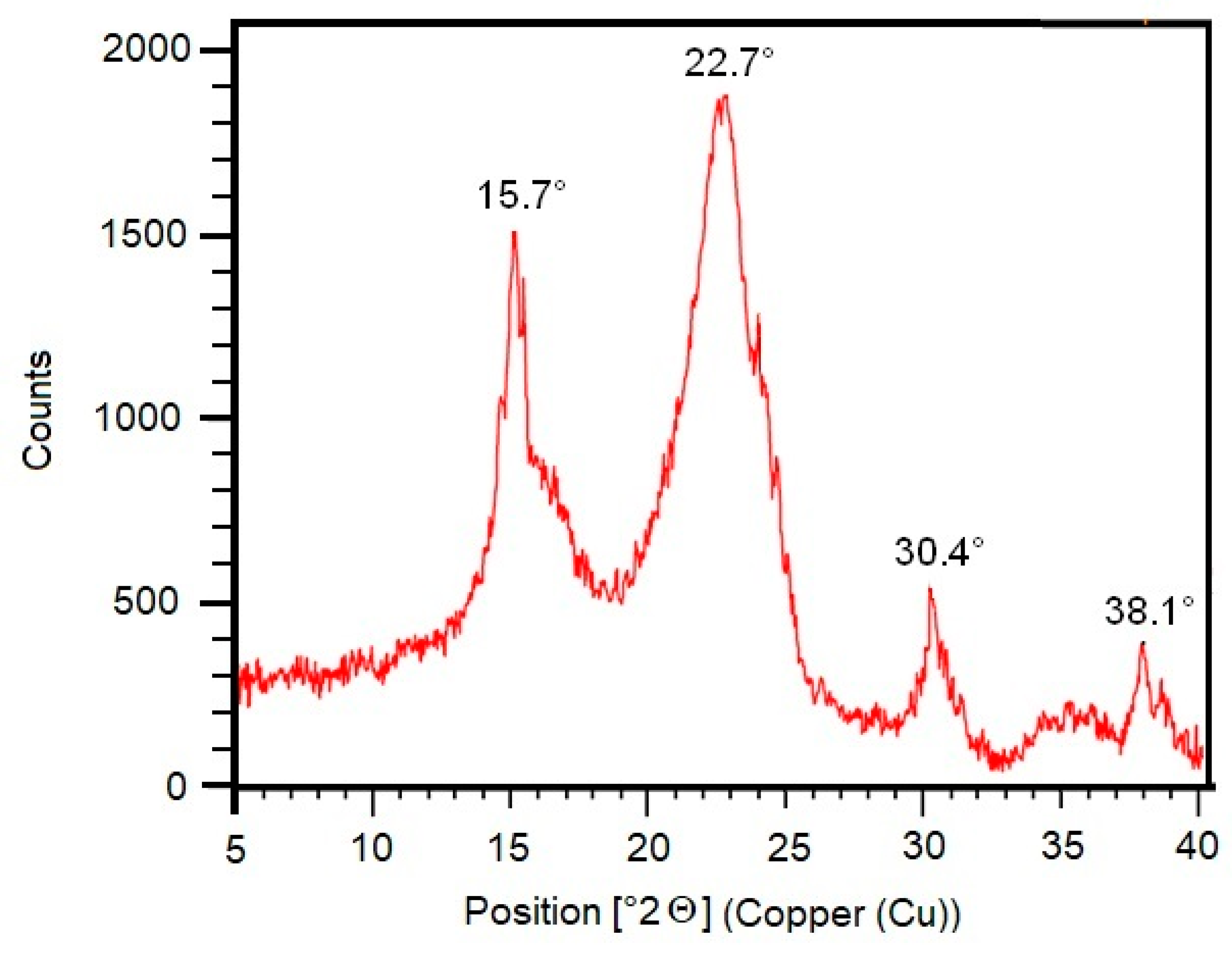
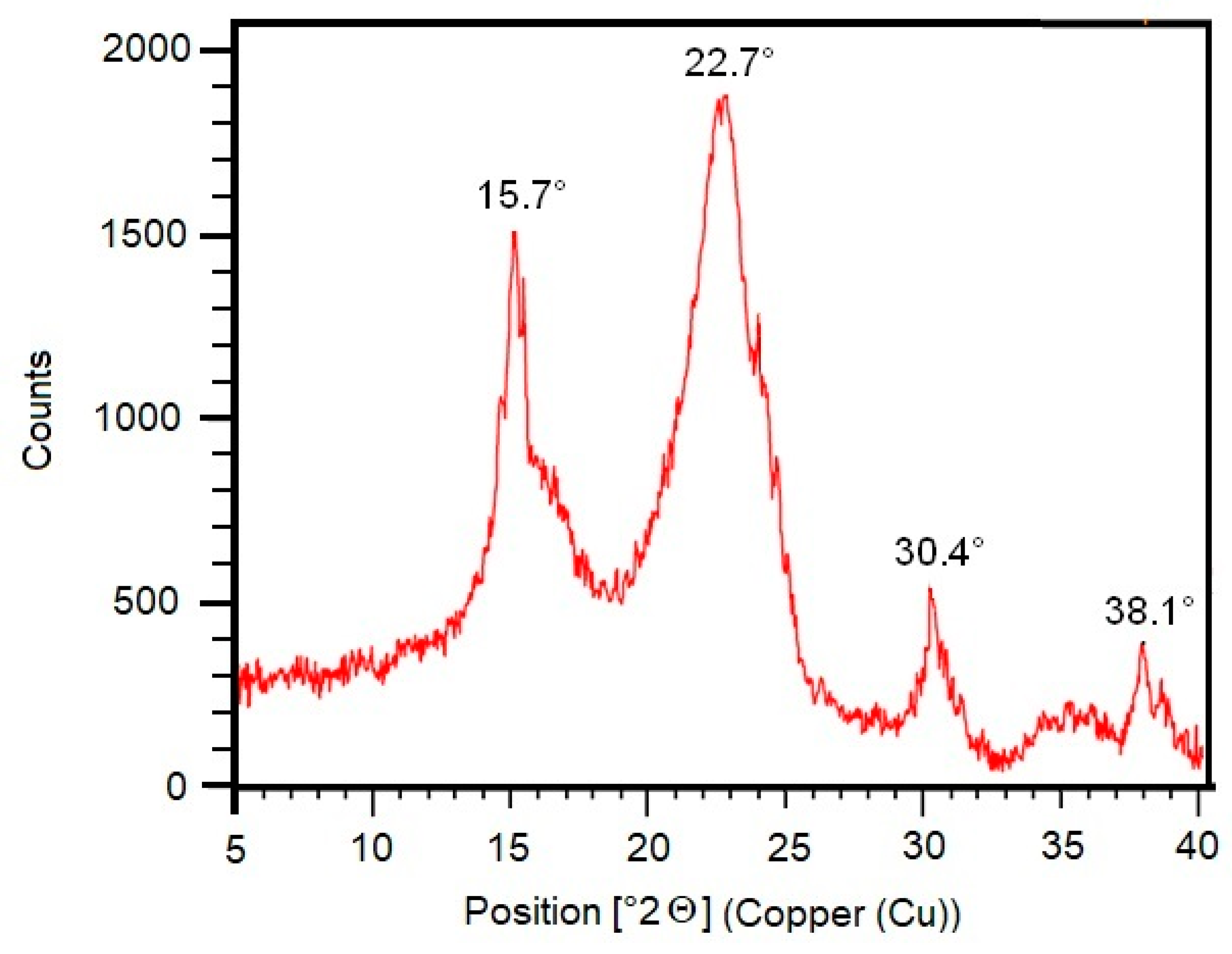
2.8. X-ray Diffractometer (XRD) Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahmad, F.; Choi, H.S.; Park, M.K. A review: Natural fiber composites selection in view of mechanical, light weight, and economic properties. Macromol. Mater. Eng. 2015, 300, 10–24. [Google Scholar] [CrossRef]
- Cheung, H.; Ho, M.; Lau, K.; Cardona, F.; Hui, D. Natural fibre-reinforced composites for bioengineering and environmental engineering applications. Compos. Part. B 2009, 40, 655–663. [Google Scholar] [CrossRef]
- Elanchezhian, C.; Vijaya Ramnath, B.; Ramakrishnan, G.; Rajendrakumar, M.; Naveenkumar, V.; Saravanakumar, M.K. Review on mechanical properties of natural fiber composites. Mater. Today Proc. 2018, 5, 1785–1790. [Google Scholar] [CrossRef]
- Kathirselvam, M.; Kumaravel, A.; Arthanarieswaran, V.P.; Saravanakumar, S.S. Isolation and characterization of cellulose fibers from Thespesia populnea barks: A study on physicochemical and structural properties. Int. J. Biol. Macromol. 2019, 129, 396–406. [Google Scholar] [CrossRef]
- Hemachandra Reddy, K.; Meenakshi Reddy, R.; Ramesh, M.; Mohana Krishnudu, D.; Madhusudhan Reddy, B.; Raghavendra Rao, H. Impact of alkali treatment on characterization of tapsi (Sterculia Urens) natural bark fiber reinforced polymer composites. J. Nat. Fibers 2019. [Google Scholar] [CrossRef]
- Amutha, V.; Senthilkumar, B. Physical, chemical, thermal, and surface morphological properties of the bark fiber extracted from Acacia concinna plant. J. Nat. Fibers 2019. [Google Scholar] [CrossRef]
- Yemele, M.C.N.; Koubaa, A.; Cloutier, A.; Soulounganga, P.; Wolcott, M. Effect of bark fiber content and size on the mechanical properties of bark/HDPE composites. Compos. Part A 2010, 41, 131–137. [Google Scholar]
- Chonsakorn, S.; Srivorradatpaisan, S. Effects of different extraction methods on some properties of water hyacinth fiber. J. Nat. Fibers 2018, 16, 1015–1025. [Google Scholar] [CrossRef]
- Baley, C. Influence of kink bands on the tensile strength of flax fibers. J. Mater. Sci. 2004, 39, 331–334. [Google Scholar] [CrossRef]
- Saravanakumaar, A.; Senthilkumar, A.; Saravanakumar, S.; Sanjay, M.R.; Khan, A. Impact of alkali treatment on physico-chemical, thermal, structural and tensile properties of Carica papaya bark fibers. Int. J. Polym. Anal. Charact. 2018, 23, 529–536. [Google Scholar] [CrossRef]
- Saravanakumar, S.; Kumaravel, A.; Nagarajan, T.; Ganesh, I. Investigation of physico-chemical properties of alkali-treated Prosopis Juliflora fibers. Int. J. Polym. Anal. Charact. 2014, 19, 37–41. [Google Scholar]
- Jayaramudu, J.; Jeevan Prasad Reddy, D.; Guduri, B.R.; Varada Rajulu, A. Properties of Natural Fabric Polyalthia Cerasoides. Fibers Polym. 2009, 10, 338–342. [Google Scholar] [CrossRef]
- Mathew, L.; Joseph, K.U.; Joseph, R. Isora fibres and their composites with natural rubber. Prog. Rubber Plast. Recycl. Technol. 2004, 20, 337–349. [Google Scholar] [CrossRef]
- Rahul, A.B.; Gautham, M.T.; Aswathy, T.R.; Indu, S.; Nair, A.S. Pharmacognostic and antibacterial activity evaluation of Acacia caesia (L.) Willd. J. Pharmacogn. Phytochem. 2020, 9, 48–54. [Google Scholar]
- Palanisamy, S.; Kalimuthu, M.; Palaniappan, M.; Alavudeen, A.; Rajini, N.; Santulli, C. Morphological Characterization of Soapbark Fibers. J. Mater. Sci. Res. Rev. 2020, 8, 19–26. [Google Scholar]
- Palanisamy, S.; Kalimuthu, M.; Palanappian, M.; Alavudeen, A.; Rajini, N.; Santulli, C. Characterization of Acacia Caesia bark fibres. In Proceedings of the ICNF2021 Conference, online, 17–19 May 2021; Fangueiro, R., Ed.; Fibrenamics: Guimaraes, Portugal. [Google Scholar]
- Sathishkumar, T.P.; Navaneethakrishnan, P.; Shankar, S. Tensile and flexural properties of snake grass natural fiber reinforced isophthalic polyester composites. Compos. Sci. Technol. 2012, 72, 1183–1190. [Google Scholar] [CrossRef]
- Van Soest, P.J.; Robertson, J.B.; Lewis, B.A. Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef]
- Spinacé, M.A.S.; Lambert, C.S.; Fermoselli, K.K.G.; De Paoli, M.A. Characterization of lignocellulosic curaua fibres. Carbohydr. Polym. 2009, 77, 47–53. [Google Scholar] [CrossRef]
- Hai, N.M.; Kim, B.S.; Lee, S. Effect of NaOH treatments on jute and coir fiber PP composites. Adv. Compos. Mater. 2009, 18, 197–208. [Google Scholar] [CrossRef]
- Bisanda, E.T.N.; Ansell, M.P. Properties of sisal-CNSL composites. J. Mater. Sci. 1992, 27, 1690–1700. [Google Scholar] [CrossRef]
- Rohen, L.A.; Neves, A.C.; Mantovani, D.D.P.; Maurício, F.C.; da Silva Vieira, J.; Pontes, L.D.A.; Monteiro, S. Hemp fiber density using the pycnometry technique. In Characterization of Minerals, Metals, and Materials; Springer: Cham, Switzerland, 2017; pp. 423–428. [Google Scholar]
- Mishra, S.; Mohanty, A.K.; Drzal, L.T.; Misra, M.; Hinrichsen, G. A review on pineapple leaf fibers, sisal fibers and their biocomposites. Macromol. Mater. Eng. 2004, 289, 955–974. [Google Scholar] [CrossRef]
- Le Gall, M.; Davies, P.; Martin, N.; Baley, C. Recommended flax fibre density values for composite property predictions. Ind. Crops Prod. 2018, 114, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Delhom, C.D.; Kelly, B.; Martin, V. Physical properties of cotton fiber and their measurement. In Cotton Fiber: Physics, Chemistry and Biology; Fang, D.D., Ed.; Springer: Cham, Switzerland, 2018; pp. 41–73. [Google Scholar]
- Satyanarayana, K.G.; Arizaga, G.G.; Wypych, F. Biodegradable composites based on lignocellulosic fibers—An overview. Prog. Polym. Sci. 2009, 34, 982–1021. [Google Scholar] [CrossRef]
- Bledzki, A.K.; Reihmane, S.; Gassan, J. Properties and modification methods for vegetable fibers for natural fiber composites. J. Appl. Polym. Sci. 1996, 59, 1329–1336. [Google Scholar] [CrossRef]
- Madhuri, K.S.; Rao, H.R.; Reddy, B.C.M.; Chandrasekhar, V. Characterization of New Cellulosic Fiber from the bark of Hardwickia Binata (Narepa). Int. J. Appl. Eng. Res. 2018, 13, 967–972. [Google Scholar]
- Charlet, K.; Eve, S.; Jernot, J.P.; Gomina, M.; Breard, J. Tensile deformation of a flax fiber. Procedia Eng. 2009, 1, 233–236. [Google Scholar] [CrossRef] [Green Version]
- Ashori, A.; Bahreini, Z. Evaluation of Calotropis gigantea as a promising raw material for fiber-reinforced composite. J. Compos. Mater. 2009, 43, 1297–1304. [Google Scholar] [CrossRef]
- Arthanarieswaran, V.P.; Kumaravel, A.; Saravanakumar, S.S. Physico-chemical properties of alkali-treated Acacia leucophloea fibers. Int. J. Polym. Anal. Charact. 2015, 20, 704–713. [Google Scholar] [CrossRef]
- Reddy, N.; Yang, Y. Natural cellulose fibers from soybean straw. Bioresource Technol. 2009, 100, 3593–3598. [Google Scholar] [CrossRef]
- Rwawiire, S.; Tomkova, B. Thermal, static, and dynamic mechanical properties of bark cloth (Ficus brachypoda) laminar epoxy composites. Polym. Compos. 2017, 38, 199–204. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Yi, X.-S.; Feng, Y. Effects of glycerin and glycerol monostearate on performance of thermoplastic starch. J. Mater. Sci. 2001, 36, 1809–1815. [Google Scholar] [CrossRef]
- Nazir, M.S.; Wahjoedi, B.A.; Yussof, A.W.; Abdullah, M.A. Eco-friendly extraction and characterization of cellulose from oil palm empty fruit bunches. Bioresource 2013, 8, 2161–2172. [Google Scholar] [CrossRef] [Green Version]
- Fatah, I.Y.A.; Abdul Khalil, H.P.S.; Hossain, M.S.; Aziz, A.A.; Davoudpour, Y.; Dungani, R.; Bhat, A. Exploration of a chemo-mechanical technique for the isolation of nanofibrillated cellulosic fiber from oil palm empty fruit bunch as a reinforcing agent in composite materials. Polymers 2014, 6, 2611–2624. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Dong, S.J.; Ma, H.H.; Zhang, B.-X.; Wang, Y.-F.; Hu, X.-M. Fractionation of corn stover into cellulose, hemicellulose and lignin using a series of ionic liquids. Ind. Crops Prod. 2015, 76, 688–696. [Google Scholar] [CrossRef]
- Sim, S.F.; Mohamed, M.; Irwan Lu, N.A.L.M.; Sarman, N.S.P.; Samsudin, S.N.S. Computer-assisted analysis of Fourier transform infrared (FTIR) spectra for characterization of various treated and untreated agriculture biomass. Bioresource 2012, 7, 5367–5380. [Google Scholar] [CrossRef] [Green Version]
- Stanzione, M.; Oliviero, M.; Cocca, M.; Errico, M.E.; Gentile, G.; Avella, M.; Lavorgna, M.; Buonocore, G.G.; Verdolotti, L. Tuning of polyurethane foam mechanical and thermal properties using ball-milled cellulose. Carbohydr. Polym. 2020, 231, 115772. [Google Scholar] [CrossRef]
- Mansikkamäki, P.; Lahtinen, M.; Rissanen, M. The conversion from cellulose I to cellulose II in NaOH mercerization performed in alcohol–water systems: An X-ray powder diffraction study. Carbohydr. Polym. 2007, 68, 35–43. [Google Scholar] [CrossRef]
- Nam, S.; French, A.D.; Condon, B.D.; Conch, M. Segal crystallinity index revisited by the simulation of X-ray diffraction patterns of cotton cellulose Iβ and cellulose II. Carbohydr. Polym. 2016, 135, 1–9. [Google Scholar] [CrossRef]
- Kathirselvam, M.; Kumaravel, A.; Arthanarieswaran, V.P.; Saravanakumar, S.S. Assessment of cellulose in bark fibers of Thespesia populnea: Influence of stem maturity on fiber characterization. Carbohydr. Polym. 2019, 212, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Saravanakumar, S.S.; Kumaravel, A.; Nagarajan, T.; Sudhakar, P.; Baskaran, R. Characterization of a novel natural cellulosic fiber from Prosopis juliflora bark. Carbohydr. Polym. 2013, 92, 1928–1933. [Google Scholar] [CrossRef]
- Elanthikkal, S.; Gopalakrishnapanicker, U.; Varghese, S.; Guthrie, J.T. Cellulose microfibres produced from banana plant wastes: Isolation and characterization. Carbohydr. Polym. 2010, 80, 852–859. [Google Scholar] [CrossRef]
- Dou, J.; Karakoç, A.; Johansson, L.S.; Hietala, S.; Evtyugin, D.; Vuorinen, T. Mild alkaline separation of fiber bundles from eucalyptus bark and their composites with cellulose acetate butyrate. Ind. Crops Prod. 2021, 165, 113436. [Google Scholar] [CrossRef]
- Dou, J.; Rissanen, M.; Ilina, P.; Mäkkylä, H.; Tammela, P.; Haslinger, S.; Vuorinen, T. Separation of fiber bundles from willow bark using sodium bicarbonate and their novel use in yarns for superior UV protection and antibacterial performance. Ind. Crops Prod. 2021, 164, 113387. [Google Scholar] [CrossRef]
- Khalid, M.Y.; Imran, R.; Arif, Z.U.; Akram, N.; Arshad, H.; Rashid, A.A.; García Márquez, F.P. Developments in Chemical Treatments, Manufacturing Techniques and Potential Applications of Natural-Fibers-Based Biodegradable Composites. Coatings 2021, 11, 293. [Google Scholar] [CrossRef]
- Baskaran, P.G.; Kathiresan, M.; Pandiarajan, P. Effect of alkali-treatment on structural, thermal, tensile properties of dichrostachys cinerea bark fiber and its composites. J. Nat. Fibers 2020, 1–17. [Google Scholar] [CrossRef]
- Peng, Y.; Nair, S.S.; Chen, H.; Farnood, R.; Yan, N.; Cao, J. Application of different bark fractions in polypropylene composites: UV and thermal stability. Polym. Compos. 2020, 41, 2198–2209. [Google Scholar] [CrossRef]
- Senthamaraikannan, P.; Saravanakumar, S.S.; Sanjay, M.R.; Jawaid, M.; Siengchin, S. Physico-chemical and thermal properties of untreated and treated Acacia planifrons bark fibers for composite reinforcement. Mater. Lett. 2019, 240, 221–224. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fibres | Density (kg/m3) | Reference |
|---|---|---|
| ACB | 1150–1200 | [16] and this work |
| Curauà | 1200 | [19] |
| Coir | 1150–1250 | [20,21] |
| Hemp | 1393 | [22] |
| Sisal | 1450 | [23] |
| Pineapple | 1440 | [23] |
| Flax | 1540 | [24] |
| Cotton | 1520 | [25] |
| Properties | Untreated ACB Fibres [15] | Alkali-Treated ACB Fibres (This Work) |
|---|---|---|
| Cellulose | 37 | 40.1 |
| Hemicellulose | 20 | 17.3 |
| Lignin | 18 | 23.5 |
| Ash | 4.3 | 5.4 |
| Moisture | 11.7 | 11.5 |
| Wax | 0.4 | 0.2 |
| Extractives | 8.6 | 2 |
| Extraction Method | Average Diameter (µm) | Standard Deviation (µm) |
|---|---|---|
| Untreated mechanically stripped | 423 | 240 |
| Treated mechanically stripped | 372 | 211 |
| Untreated further separated | 127 | 25 |
| Treated further separated | 123 | 22 |
| Avg. Value | Std. Deviation | Variance | ||
|---|---|---|---|---|
| Untreated fibres | Ultimate load (N) | 10.8 | 5.21 | 48.31 |
| Elongation (%) | 3.4 | 1.0 | 30.75 | |
| 5% NaOH treated fibres | Ultimate load (N) | 11.14 | 4.56 | 20.82 |
| Elongation (%) | 4.25 | 0.80 | 0.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivasubramanian, P.; Kalimuthu, M.; Palaniappan, M.; Alavudeen, A.; Rajini, N.; Santulli, C. Effect of Alkali Treatment on the Properties of Acacia Caesia Bark Fibres. Fibers 2021, 9, 49. https://0-doi-org.brum.beds.ac.uk/10.3390/fib9080049
Sivasubramanian P, Kalimuthu M, Palaniappan M, Alavudeen A, Rajini N, Santulli C. Effect of Alkali Treatment on the Properties of Acacia Caesia Bark Fibres. Fibers. 2021; 9(8):49. https://0-doi-org.brum.beds.ac.uk/10.3390/fib9080049
Chicago/Turabian StyleSivasubramanian, Palanisamy, Mayandi Kalimuthu, Murugesan Palaniappan, Azeez Alavudeen, Nagarajan Rajini, and Carlo Santulli. 2021. "Effect of Alkali Treatment on the Properties of Acacia Caesia Bark Fibres" Fibers 9, no. 8: 49. https://0-doi-org.brum.beds.ac.uk/10.3390/fib9080049