
Molecular Assay Development to Monitor the Kinetics of Viable Populations of Two Biocontrol Agents, Bacillus subtilis QST 713 and Gliocladium catenulatum J1446, in the Phyllosphere of Lettuce Leaves


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. The Biocontrol Agents Used: B. subtilis QST 713 and G. catenulatum J1446
2.2. Cell Suspensions and Viability of Biocontrol Population Treatments
2.3. PMAxx™ Treatment
2.4. DNA Extraction and qPCR
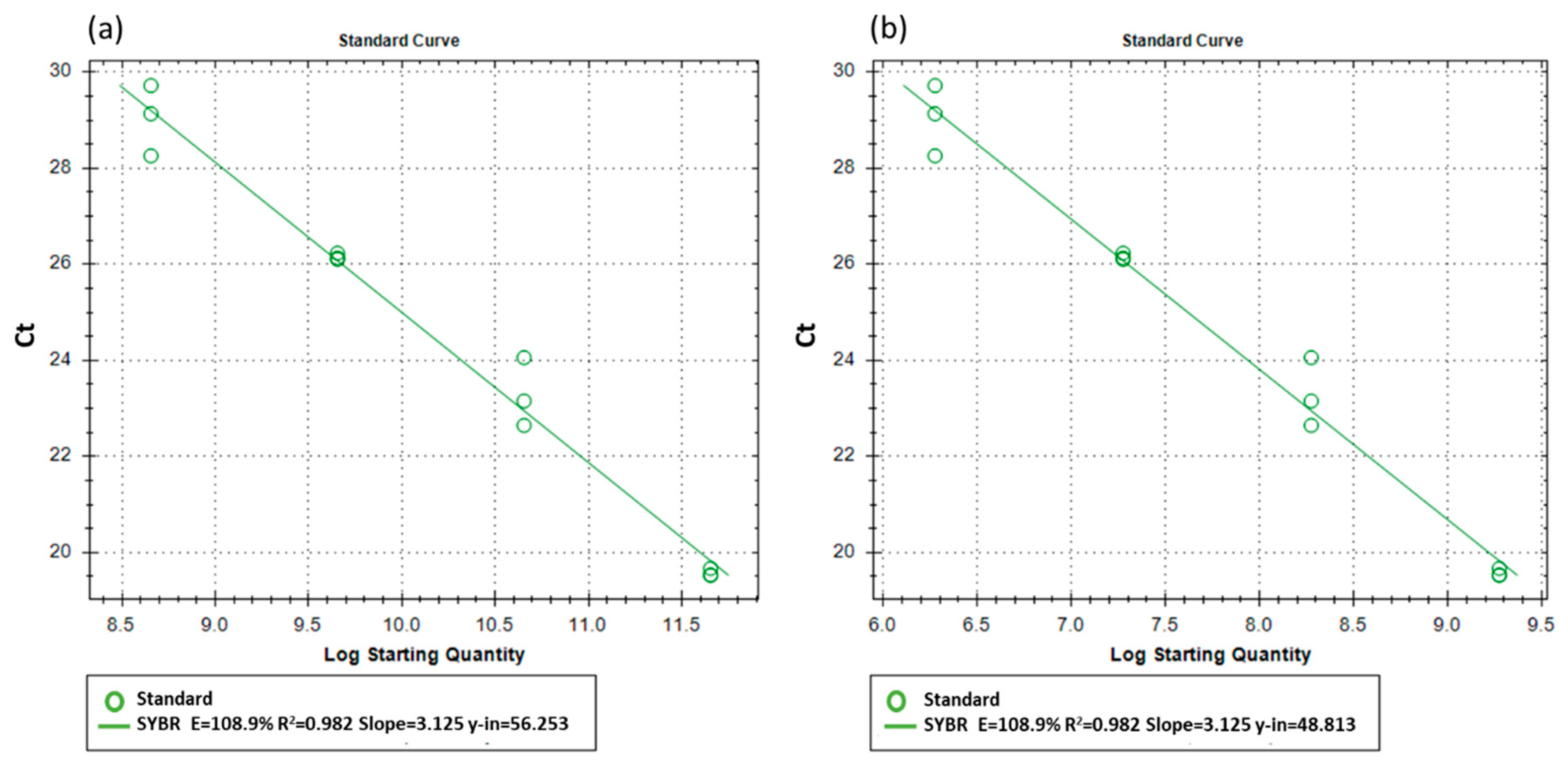
2.5. Standard Curves of Cycle Threshold to Copy Number of DNA and Viable Population
2.6. Optimisation of the PMAxx-PCR Assays
2.7. Testing for Viable BCA Quantification with the Optimised PMAxxTM-qPCR from the Phyllosphere of Lettuce
- (1)
- Plant propagation and selection; plants (Lactuca sativa cv. Carter) were sown, grown, and selected for being pest and disease-free and healthy, with a minimum of six leaves.
- (2)
- BCA cultivation and concentration calibration; plate counts were used to determine the concentration of the cultivated BCAs and were adjusted as necessary to obtain 8 log10 CFUs/mL for B. subtilis, and 8 log10 spores/mL for G. catenulatum.
- (3)
- Plant treatment; plants were sprayed with the appropriate BCA as a fine droplet setting until just before run-off.
- (4)
- Plant drying; after treatment plants were allowed to dry for 1 h in the glasshouse, and then placed into their designated glasshouse and/or polytunnel.
- (5)
- Sampling was done on days 0 (1 h after BCA application), 2, 4, 6, 8 and 10 after spraying. On each sampling day the oldest leaf was collected from five pre-determined plants (one leaf per plant), and immediately pooled and placed into a falcon tube containing maximum recovery diluent (Sigma).
- (6)
- Surface washing, filtration, and cell pellet collection; the leaves were soaked in maximum recovery diluent until full, sealed, and shaken on a rotary shaker at 100 rpm for 30 min at 10 °C. The contents were filtered with a wet muslin cloth (four layers), and the cells were pelleted by centrifugation at 2000× g for 15 min at 4 °C. The supernatant was decanted, and the cell pellet supplemented up to 500 μL with maximum recovery diluent solution, and then transferred into a 1.5 mL Eppendorf tube for storage at 4 °C.
- (7)
- PMAxx™ treatment (25 μM for B. subtilis; 50 μM for G. catenulatum) and the DNA extraction and qPCR methods were previously described.
3. Results
3.1. Effect of PMAxx™ on Blocking DNA Amplification of Dead BCA Cells
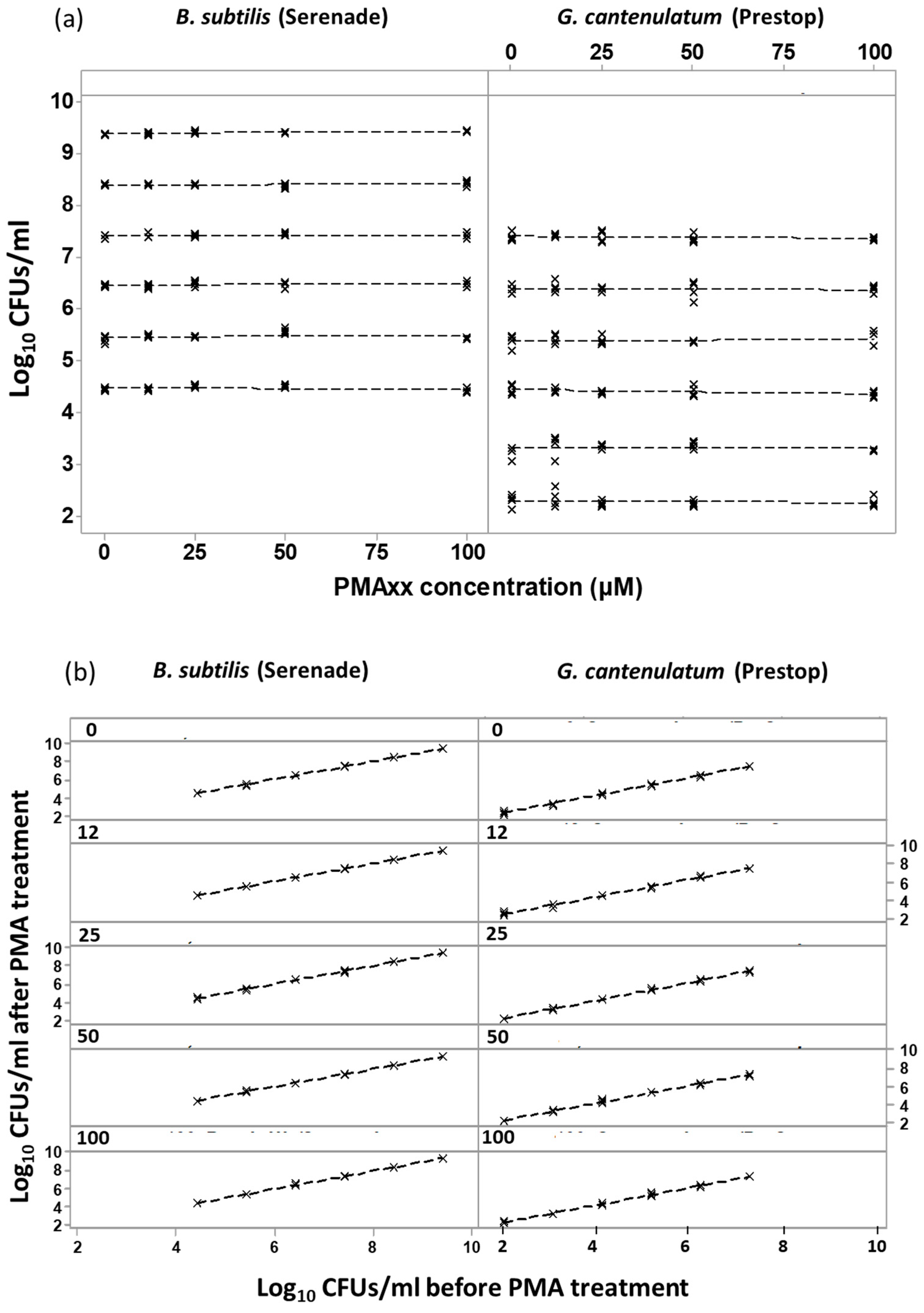
3.2. PMAxx™ Concentration Response at Different Numbers of Total log10 Dead Cells
3.3. Assays for Testing PMAxx™ Concentration on In Vitro Viability Counts of the Two BCAs
3.4. Sensitivity Range of PMAxxTM-qPCR on Blocking DNA from Dead Cells
3.5. Temporal Fluxes in Phyllosphere Quantification of the Two BCAs on Lettuce Leaves Using the PMAxxTM-qPCR Technique
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, K.P.; Handelsman, J.; Goodman, R.M. Modeling dose-response relationships in biological control: Partitioning host responses to the pathogen and biocontrol agent. Phytopathology 1997, 7, 720–729. [Google Scholar] [CrossRef] [Green Version]
- Turano, A.; Pirali, F. Quantification Methods in Microbiology. In Laboratory Diagnosis of Infectious Diseases; Springer: Berlin/Heidelberg, Germany, 1988. [Google Scholar]
- Papadimitriou, K.; Alegria, A.; Bron, P.A.; De Angelis, M.; Gobbetti, M.; Kleerebezem, M.; Lemos, J.A.; Linares, D.M.; Ross, P.; Stanton, C.; et al. Stress Physiology of Lactic Acid Bacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 837–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Deng, Y.; Peters, B.M.; Li, L.; Li, B.; Chen, L.; Xu, Z.; Shirtliff, M.E. Transcriptomic analysis on the formation of the viable putative non-culturable state of beer-spoilage Lactobacillus acetotolerans. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sanzani, S.M.; Li Destri Nicosia, M.G.; Faedda, R.; Cacciola, S.O.; Schena, L. Use of quantitative PCR detection methods to study biocontrol agents and phytopathogenic fungi and oomycetes in environmental samples. J. Phytopath. 2014, 162, 1–13. [Google Scholar] [CrossRef]
- Silva, D.M.; Domingues, L. On the track for an efficient detection of Escherichia coli in water: A review on PCR-based methods. Ecotox. Environ. Safety 2015, 113, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun-Kiewnick, A.; Lehmann, A.; Rezzonico, F.; Wend, C.; Smits, T.H.; Duffy, B. Development of species-, strain- and antibiotic biosynthesis-specific quantitative PCR assays for Pantoea agglomerans as tools for biocontrol monitoring. J. Microbiol. Methods 2012, 90, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Pujol, M.; Badosa, E.; Manceau, C.; Montesinos, E. Assessment of the environmental fate of the biological control agent of fire blight, Pseudomonas fluorescens EPS62e, on apple by culture and real-time PCR methods. Appl. Environ. Microbiol. 2006, 72, 2421–2427. [Google Scholar] [CrossRef] [Green Version]
- Spotts, R.A.; Seifert, K.A.; Wallis, K.M.; Sugar, D.; Xiao, C.L.; Serdani, M.; Henriquez, J.L. Description of Cryptosporiopsis kienholzii and species profiles of Neofabraea in major pome fruit growing districts in the Pacific Northwest USA. Mycol. Res. 2009, 113, 1301–1311. [Google Scholar] [CrossRef]
- Fittipaldi, M.; Nocker, A.; Codony, F. Progress in understanding preferential detection of live cells using viability dyes in combination with DNA amplification. J. Microbiol. Methods 2012, 91, 276–289. [Google Scholar] [CrossRef]
- Nocker, A.; Cheung, C.-Y.; Camper, A.K. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J. Microbiol. Methods 2006, 67, 310–320. [Google Scholar] [CrossRef]
- Nocker, A.; Camper, A.K. Novel approaches toward preferential detection of viable cells using nucleic acid amplification techniques. FEMS Microbiol. Lett. 2009, 291, 137–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nocker, A.; Sossa, K.E.; Camper, A.K. Molecular monitoring of disinfection efficacy using propidium monoazide in combination with quantitative PCR. J. Microbiol. Methods 2007, 70, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Elizaquível, P.; Sánchez, G.; Aznar, R. Quantitative detection of viable foodborne E. coli O157: H7, Listeria monocytogenes and Salmonella in fresh-cut vegetables combining propidium monoazide and real-time PCR. Food Cont. 2012, 25, 704–708. [Google Scholar] [CrossRef]
- Crespo-Sempere, A.; Estiarte, N.; Marín, S.; Sanchis, V.; Ramos, A.J. Propidium monoazide combined with real-time quantitative PCR to quantify viable Alternaria spp. contamination in tomato products. Int. J. Food Microbiol. 2013, 165, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Soto-Muñoz, L.; Teixidó, N.; Usall, J.; Viñas, I.; Crespo-Sempere, A.; Torres, R. Development of PMA real-time PCR method to quantify viable cells of Pantoea agglomerans CPA-2, an antagonist to control the major postharvest diseases on oranges. Int. J. Food Microbiol. 2014, 180, 49–55. [Google Scholar] [CrossRef]
- Soto-Muñoz, L.; Torres, R.; Usall, J.; Viñas, I.; Dashevskaya, S.; Teixidó, N. Environmental monitoring of the biocontrol agent Pantoea agglomerans CPA-2 applied to citrus fruit at preharvest. Annals Appl. Biol. 2015, 167, 250–261. [Google Scholar] [CrossRef]
- Hertwig, C.; Steins, V.; Reineke, K.; Rademacher, A.; Klocke, M.; Rauh, C.; Schluter, O. Impact of surface structure and feed gas composition on Bacillus subtilis endospore inactivation during direct plasma treatment. Front. Microbiol. 2015, 6, 774. [Google Scholar] [CrossRef] [PubMed]
- Paavanen-Huhtala, S.; Avikainen, H.; Yli-Mattila, T. Development of strain-specific primers for a strain of Gliocladium catenulatum used in biological control. Eur. J. Plant Pathol. 2000, 106, 187–198. [Google Scholar] [CrossRef]
- Barbau-Piednoir, E.; Mahillon, J.; Pillyser, J.; Coucke, W.; Roosens, N.H.; Botteldoorn, N. Evaluation of viability-qPCR detection system on viable and dead Salmonella serovar Enteritidis. J. Microbiol. Methods 2014, 103, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Tut, G. Optimising the Use of Biocontrol Agents to Improve the Control of Botrytis cinerea in Key Vegetable and Fruit Crops. Ph.D. Thesis, Cranfield University, Cranfield, UK, 2020. [Google Scholar]
- Trias, R.; Baneras, L.; Montesinos, E.; Badosa, E. Lactic acid bacteria from fresh fruit and vegetables as biocontrol agents of phytopathogenic bacteria and fungi. Int. Microbiol. 2008, 11, 231–236. [Google Scholar] [PubMed] [Green Version]
- Larena, I.; Melgarejo, P. Development of a new strategy for monitoring Epicoccum nigrum 282, a biological control agent used against brown rot caused by Monilinia spp. in peaches. Postharvest Biol. Technol. 2009, 54, 63–71. [Google Scholar] [CrossRef]
- Daranas, N.; Bonaterra, A.; Frances, J.; Cabrefiga, J.; Montesinos, E.; Badosa, E. Monitoring viable cells of the biological control agent Lactobacillus plantarum PM411 in aerial plant surfaces by means of a strain-specific viability quantitative PCR method. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [Green Version]
- Emerson, J.B.; Adams, R.I.; Román, C.M.B.; Brooks, B.; Coil, D.A.; Dahlhausen, K.; Ganz, H.H.; Hartmann, E.M.; Hsu, T.; Justice, N.B. Schrödinger’s microbes: Tools for distinguishing the living from the dead in microbial ecosystems. Microbiome 2017, 5, 86. [Google Scholar] [CrossRef]
- Pan, Y.; Breidt, F. Enumeration of viable Listeria monocytogenes cells by real-time PCR with propidium monoazide and ethidium monoazide in the presence of dead cells. Appl. Environ. Microbiol. 2007, 73, 8028–8031. [Google Scholar] [CrossRef] [Green Version]
- Yáñez, M.A.; Nocker, A.; Soria-Soria, E.; Múrtula, R.; Martínez, L.; Catalán, V. Quantification of viable Legionella pneumophila cells using propidium monoazide combined with quantitative PCR. J. Microbiol. Methods 2011, 85, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.J.; Bentham, R.H.; Ross, K.E. Limitations of using propidium monoazide with qPCR to discriminate between live and dead Legionella in biofilm samples. Microbiol. Insights 2014, 7, 15–24. [Google Scholar] [CrossRef]
- Nkuipou-Kenfack, E.; Engel, H.; Fakih, S.; Nocker, A. Improving efficiency of viability-PCR for selective detection of live cells. J. Micrbiol. Methods 2013, 93, 20–24. [Google Scholar] [CrossRef]
- Martinon, A.; Cronin, U.; Quealy, J.; Stapleton, A.; Wilkinson, M. Swab sample preparation and viable real-time PCR methodologies for the recovery of Escherichia coli, Staphylococcus aureus or Listeria monocytogenes from artificially contaminated food processing surfaces. Food Cont. 2012, 24, 86–94. [Google Scholar] [CrossRef]
- Martin, B.; Raurich, S.; Garriga, M.; Aymerich, T. Effect of amplicon length in propidium monoazide quantitative PCR for the enumeration of viable cells of Salmonella in cooked ham. Food Anal. Meth. 2013, 6, 683–690. [Google Scholar] [CrossRef]
- Seinige, D.; Krischek, C.; Klein, G.; Kehrenberg, C. Comparative analysis and limitations of ethidium monoazide and propidium monoazide treatments for the differentiation of viable and nonviable Campylobacter cells. Appl. Environ. Microbiol. 2014, 80, 2186–2192. [Google Scholar] [CrossRef] [Green Version]
- Rudi, K.; Moen, B.; Drømtorp, S.M.; Holck, A.L. Use of ethidium monoazide and PCR in combination for quantification of viable and dead cells in complex samples. Appl. Environ. Microbiol. 2005, 71, 1018–1024. [Google Scholar] [CrossRef] [Green Version]
- Sinskey, T.; Silverman, G. Characterization of injury incurred by Escherichia coli upon freeze-drying. J. Bacteriol. 1970, 101, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Abadias, M.; Teixido, N.; Usall, J.; Vinas, I.; Magan, N. Solute stresses affect growth patterns, endogenous water potentials and accumulation of sugars and sugar alcohols in cells of the biocontrol yeast Candida sake. J. Appl. Microbiol. 2000, 89, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Abadias, M.; Teixido, N.; Usall, J.; Vinas, I.; Magan, N. Improving water stress tolerance of the biocontrol yeast Candida sake grown in molasses-based media by physiological manipulation. Can. J. Microbiol. 2001, 47, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Abadias, M.; Teixido, N.; Usall, J.; Vinas, I. Liquid formulation of the postharvest biocontrol agent Candida sake CPA-1 in isotonic solutions. Phytopathology 2003, 93, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Knight, I.T. Molecular genetic methods for detection and identification of viable but non-culturable microorganisms. In Nonculturable Microorganisms in the Environment; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar]
- Dashti, A.A.; Jadaon, M.M.; Abdulsamad, A.M.; Dashti, H.M. Heat treatment of bacteria: A simple method of DNA extraction for molecular techniques. Kuwait Med. J. 2009, 41, 117–122. [Google Scholar]
- Desfossés-Foucault, É.; Dussault-Lepage, V.; Le Boucher, C.; Savard, P.; Lapointe, G.; Roy, D. Assessment of probiotic viability during Cheddar cheese manufacture and ripening using propidium monoazide-PCR quantification. Front. Microbiol. 2012, 3, 350. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tut, G.; Magan, N.; Brain, P.; Xu, X. Molecular Assay Development to Monitor the Kinetics of Viable Populations of Two Biocontrol Agents, Bacillus subtilis QST 713 and Gliocladium catenulatum J1446, in the Phyllosphere of Lettuce Leaves. Biology 2021, 10, 224. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10030224
Tut G, Magan N, Brain P, Xu X. Molecular Assay Development to Monitor the Kinetics of Viable Populations of Two Biocontrol Agents, Bacillus subtilis QST 713 and Gliocladium catenulatum J1446, in the Phyllosphere of Lettuce Leaves. Biology. 2021; 10(3):224. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10030224
Chicago/Turabian StyleTut, Gurkan, Naresh Magan, Philip Brain, and Xiangming Xu. 2021. "Molecular Assay Development to Monitor the Kinetics of Viable Populations of Two Biocontrol Agents, Bacillus subtilis QST 713 and Gliocladium catenulatum J1446, in the Phyllosphere of Lettuce Leaves" Biology 10, no. 3: 224. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10030224