
An “Omic” Overview of Fragile X Syndrome


Department of Biochemistry and Functional Genomics, Faculty of Medicine and Health Sciences, Université de Sherbrooke and Centre de Recherche du CHUS, CIUSSS de l’Estrie-CHUS, Sherbrooke, QC J1H 5H4, Canada
*
Author to whom correspondence should be addressed.
Biology 2021, 10(5), 433; https://0-doi-org.brum.beds.ac.uk/10.3390/biology10050433
Submission received: 24 March 2021
/
Revised: 1 May 2021
/
Accepted: 8 May 2021
/
Published: 13 May 2021
(This article belongs to the Special Issue RNA-Binding Proteins: Function, Dysfunction and Disease)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Fragile X syndrome (FXS) is a neurodevelopmental disorder and remains the most frequent inherited cause of intellectual disability. Fragile X patients are at great risk to develop behavior problems including autism, anxiety, aggressivity and hyperactivity. FXS results from a mutation leading to the absence of a single protein, the fragile X mental retardation protein (FMRP). Most studies aiming to understand the physiopathology of FXS are centered on the effect of its absence on protein synthesis. However, besides protein synthesis regulation, FMRP has several other functions that need to be understood and could play a significant role in FXS. The goal of the present work is to review some of these functions and to put them into perspective in order to get a more comprehensive understanding of FXS physiopathology.
Abstract
Fragile X syndrome (FXS) is a neurodevelopmental disorder associated with a wide range of cognitive, behavioral and medical problems. It arises from the silencing of the fragile X mental retardation 1 (FMR1) gene and, consequently, in the absence of its encoded protein, FMRP (fragile X mental retardation protein). FMRP is a ubiquitously expressed and multifunctional RNA-binding protein, primarily considered as a translational regulator. Pre-clinical studies of the past two decades have therefore focused on this function to relate FMRP’s absence to the molecular mechanisms underlying FXS physiopathology. Based on these data, successful pharmacological strategies were developed to rescue fragile X phenotype in animal models. Unfortunately, these results did not translate into humans as clinical trials using same therapeutic approaches did not reach the expected outcomes. These failures highlight the need to put into perspective the different functions of FMRP in order to get a more comprehensive understanding of FXS pathophysiology. This work presents a review of FMRP’s involvement on noteworthy molecular mechanisms that may ultimately contribute to various biochemical alterations composing the fragile X phenotype.
1. Introduction
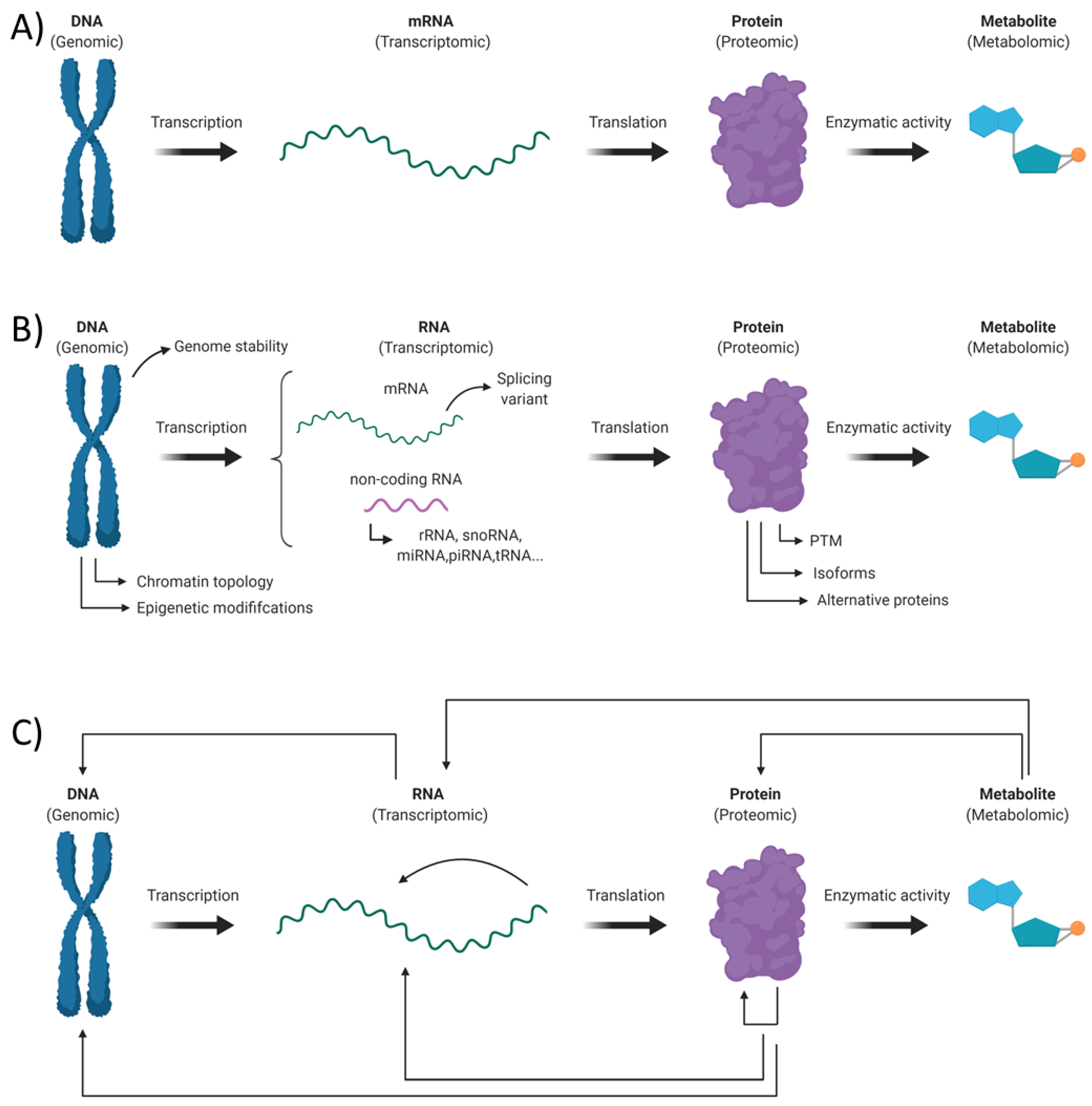
The “Omic” term refers to the wide range of high throughput experimental approaches aiming to get a holistic depiction of the different biomolecules within a living organism. These techniques intend to get a better understanding of systems biology throughout a comprehensive overview of the genes (genomic), RNA (transcriptomic), proteins (proteomic) and metabolites (metabolomic) making up a cell or a tissue. However, when performing such studies, one should avoid the mistake of only getting a simplistic “Omic” portrait of its biological sample (Figure 1A). Indeed, an aware experimenter should take in consideration the large and inherent diversity within each class of biological molecules and the complex nature of the different interactions between them (Figure 1B,C).
Fragile X syndrome (FXS) is an X-linked neurodevelopmental disorder with an approximative prevalence of 1/4000 in males and 1/8000 in females [1]. FXS is the leading inherited cause of intellectual disability (ID) and the most prevalent monogenic cause of autism spectrum disorders (ASD). The severity of ID varies from one individual to another, being usually moderate to severe in men while it is most often mild to borderline in women. It is estimated that between 30% and 50% of patients with FXS will present ASD. Fragile X syndrome is also characterized by a broad spectrum of challenging behaviors (anxiety, aggression, self-injury, attention deficit disorder (ADHD, social withdrawal), physical (facial dysmorphia, macroorchidism, joints hypermobility) and self-limited medical problems (recurrent otitis media, seizures). Furthermore, all FXS-related comorbidities are found with variable penetrance, resulting in a large phenotypic heterogeneity across this population [2,3,4,5].
Fragile X syndrome typically originates from the dynamic expansion of CGG trinucleotide repeats found in the 5′ UTR of the FMR1 gene (Xq27.3) [6]. A full mutation allele possesses more than 200 repeats, which results in the transcriptional silencing of the gene throughout an epigenetic mechanism and ultimately in the loss of expression of the fragile X mental retardation protein (FMRP) [6,7]. Mosaicism is commonly observed in the fragile X population, with a prevalence ranging from 12–41% in males [8,9]. Repeats-size and methylation mosaicisms have been described in FXS and are both associated with a detectable FMRP expression, a higher IQ and milder behavioral problems [10,11,12,13] (Figure 2). X chromosome inactivation ratio in fragile X females (proportion of cells that have inactivated the fully mutated X chromosome) have also been correlated with a better prognosis [10].
FMRP is widely expressed across the body but to a higher degree in the brain and gonads [14,15]. FMRP possesses both nuclear localization and exportation signals, two Agenet domains, as well as two hnRNP homology domains (KH) and one arginine–glycine–glycine box (RGG) [16]. The last three domains provide FMRP RNA binding capacity. FMRP is able to recognize several RNA motifs such as pseudoknots’ structure, SoSLIP and G-quartets [17,18,19], as well as specific RNA sequences [20]. The RNA binding capacity of FMRP is central to its molecular function. Indeed, missense mutations in either KH domains impair FMRP ability to bind RNA and are associated with many typical FXS phenotypes, including ID, facial dysmorphia and macroorchidism [21,22]. Despite the fact that FMRP is involved in many stages of RNA metabolism, translational regulation remains its foremost function [23,24,25].
FMRP functions are essential for normal neurodevelopment, as illustrated by the many neuroanatomical defects harbored by fragile X patients. Indeed, abnormalities in white and grey matter volume of different brain regions have been observed in both young and adult patients [26,27,28]. Furthermore, post-mortem studies of fragile X brains have revealed an aberrant spine morphology, characterized by longer, thinner and denser dendritic spines [29,30]. Synaptic plasticity is impaired in the absence of FMRP and seems to be related to cortex hyperexcitability observed in FXS patients and several animal models [31,32,33]. This hyperexcitability seems to be the result of an enhanced long-term depression (LTD) and reduced long-term potentiation (LTP), the contribution of both mechanisms being different according to brain regions [34]. In some patients, the absence of FMRP may also affect other classic neurochemicals pathways, including the dopaminergic, cholinergic and serotonergic-mediated pathways [35,36].
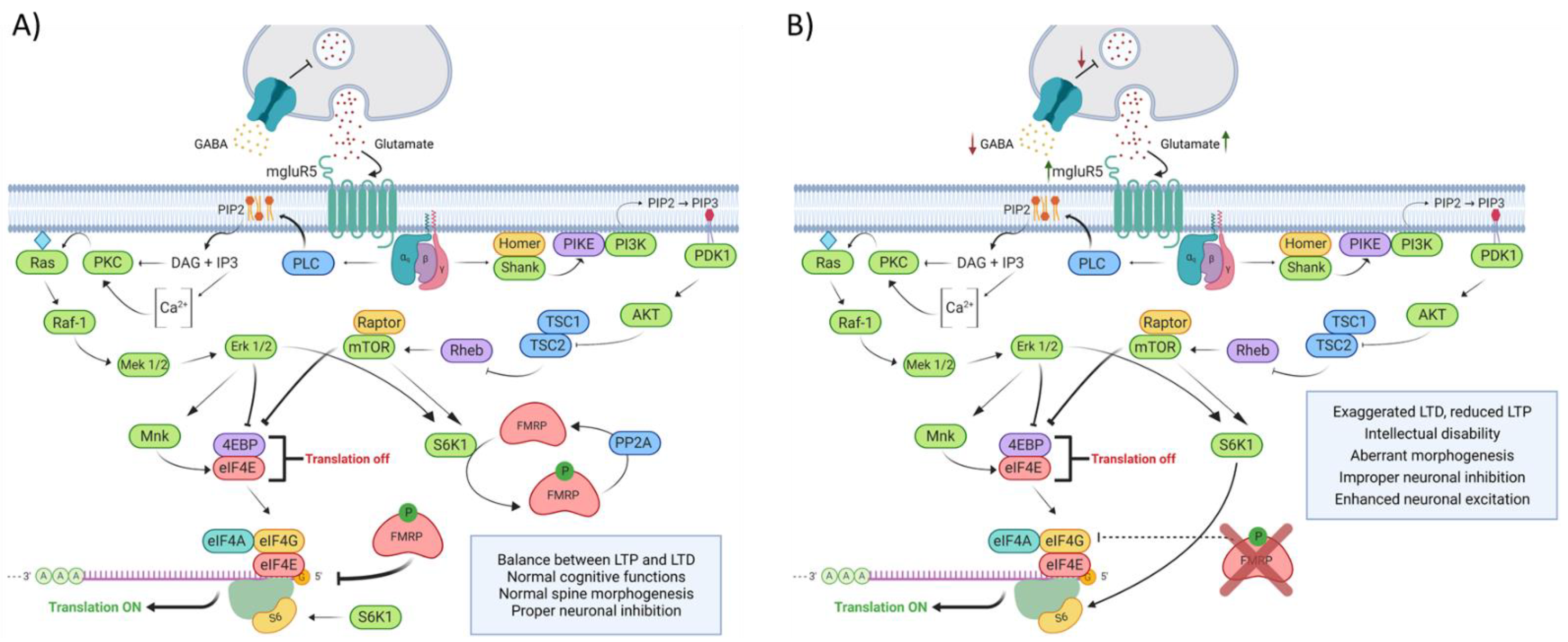
The preclinical research conducted over the past 20 years has mainly focused on the excitatory glutamate transmission mediated by group 1 (Gp1) metabotropic glutamate receptor (mGluR1 and mGluR5) and the γ-aminobutyric acid (GABA) related inhibition to link the lack of FMRP to the mechanisms underlaying FXS physiopathology (Figure 3). The hypothesis based upon those observations are centered around FMRP’s ability to regulate the translation of its targeted mRNA. In this conjecture, FMRP would act as a chief regulator of Gp1 mGluR signaling by mediating the translation of proteins involved in its signal transduction and by functionally opposing the pro-translational effects of Gp1 mGluR stimulation [24]. The upregulation of Gp1 mGluR signaling results in aberrant LTD and LTP found in the Fmr1 KO mouse hippocampus [37,38]. Deficits in GABAergic inhibition are also found in the FXS mouse model. Indeed, FMRP has been shown to regulate the translation of some GABA receptors subunits and many enzymes involved in GABA metabolism and transport [39,40,41,42]. Taken together, those alterations are thought to create an imbalance between excitatory and inhibitory neurotransmission and are believed to underline the cognitive and behavioral phenotypes observed in FXS.
This model of FXS physiopathology has been corroborated over the years by a plethora of animal model-driven studies. Indeed, the restoration of GABA and glutamate neurotransmission in Fmr1 KO mice have repeatedly been associated with the rescue of FX biochemical and behavioral phenotypes [43,44,45,46,47,48,49,50]. Unfortunately, the hype carried out by those preclinical investigations did not translate into human, as clinical trials aimed to inhibit mGluR5 or to enhance the GABAergic system did not completely fulfill the expected outcomes [51,52,53,54,55,56,57,58,59]. Lessons can be learned from this inability to translate the success generated in the mouse model to the fragile X patient. Firstly, a better understanding of human physiopathology is needed. Given the accumulation of evidence regarding the limitation of the KO mouse to fully replicate human phenotype, studies aiming to fulfill this task must focus on human subjects and models [60,61]. Secondly, one should “think outside of the ribosome” when searching for new mechanisms underlaying FXS physiopathology. The bulk of the research addressing this topic has solely considered FMRP as a translational regulator. The assumption that FXS is merely a disease of erroneous translation compose an oversimplification of the broad diversity of biochemicals and cellular alterations composing the fragile X phenotype. Finally, one should always keep in mind the large phenotypic heterogeneity found across the FXS population.
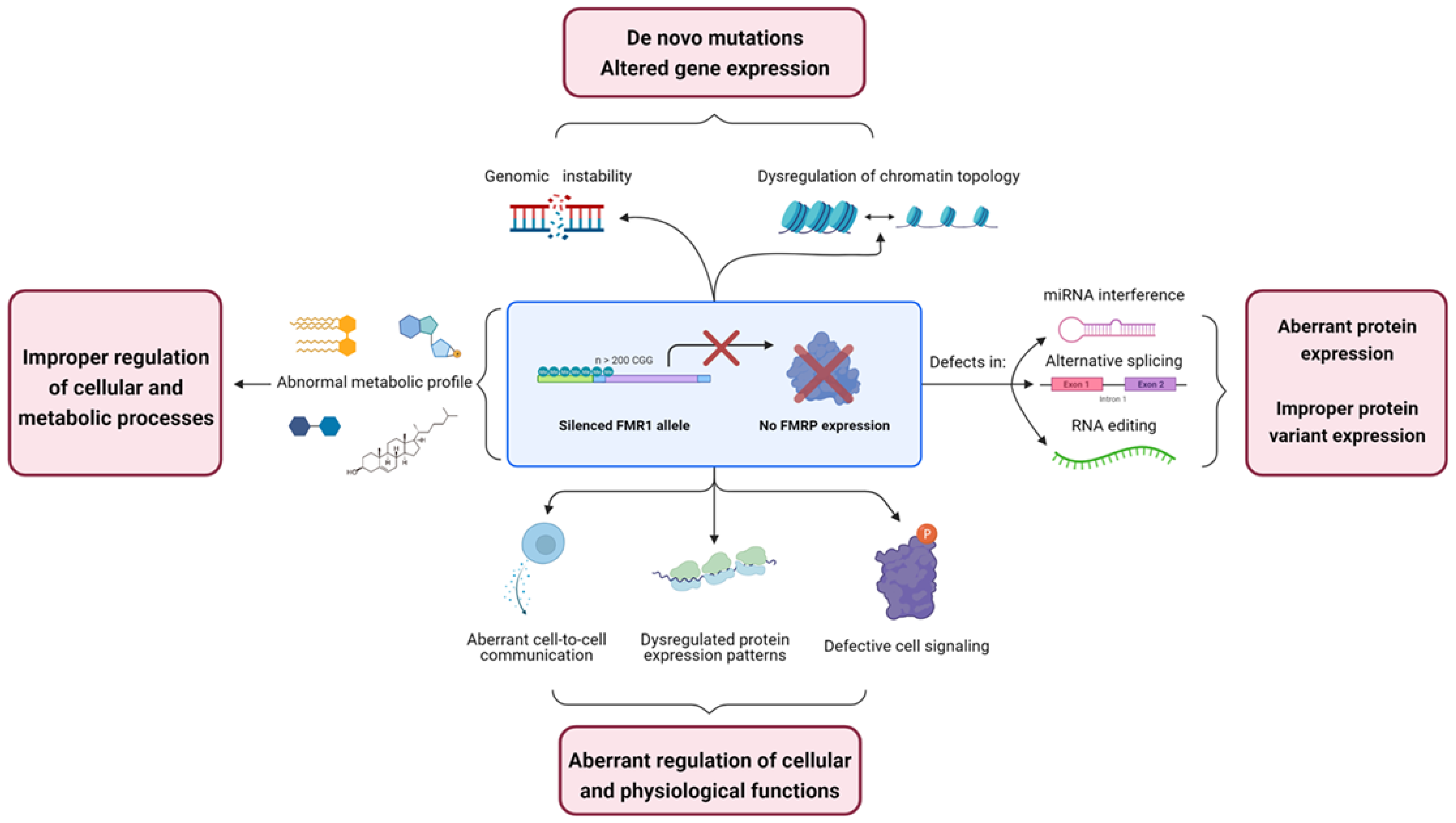
This work proposes an overview of some of the numerous molecular mechanisms in which FMRP is involved and the multidimensional nature of the biochemicals defects found in fragile X (Figure 4). This will be accomplished by reviewing some of the different alteration observable in FXS in an “Omic-dependent” manner. Furthermore, this review will focus, as much as possible, on research conducted in human subjects or models.
2. Genomic Alterations in Fragile X Syndrome
Even though the vast majority is cytoplasmic, a certain proportion of FMRP is located in the nucleus. Some isoforms in which the nuclear export signal is spliced out are indeed found predominantly in this cellular compartment [62,63,64]. Those observations initially raised the hypothesis that FMRP may have actual nuclear function.
2.1. FMRP Is Involved in Genomic Stability Maintenance
One of the first reports of FMRP genomic functions came from the study of Alpatov et al., in which they brought strong evidence of FMRP implication in DNA damage response (DDR) [65]. They discovered that FMRP, through its Agenet domains, binds the chromatin through the methylated tails of histones H3. Mouse embryonic fibroblasts (MEF) treated with replication stress agent showed a FMRP-dependent induction of histone H2A variant H2A.X phosphorylation (γH2A.X), a core component of the DNA repair complex [66]. They also showed that under these conditions, more FMRP is located in the nucleus and that some of it colocalize with γH2A.X. FMRP transitory expression can rescue γH2A.X induction in HeLa and MEF cells, but mutations impairing Agenet domain binding to the chromatin prove to be less efficient in the matter. The precise role of FMRP in DDR remains, nonetheless, to be fully elucidated.
Evidence of the drosophila homolog of FMRP (dFMRP) involvement in the Piwi-interacting RNA (piRNA) pathway have also been reported [67,68]. The piRNA pathway is involved in the transcriptional silencing and enzymatic degradation of transposable elements, which have the ability to replicate and insert into new loci [69]. The piRNA mediated inhibition is crucial for the maintenance of genome integrity, as transposition events can lead to DNA breaks and improper recombination.
Taken together, those studies point toward a role for FMRP in the maintenance of genomic stability, which further suggests that fragile X patients may be subject to an abnormal increase of mutations caused by an accumulation of DNA damage and improper transposition events. This phenomenon can have a deleterious effect on long-lasting cells such as neurons and potentially have repercussion on the wider nervous circuit in which affected neurons are connected.
2.2. Alteration of Chromatin Topology in FXS
Regulation of the genome three-dimensional structure, also known as chromatin topology, is crucial for the spatiotemporal control of gene expression. This tightly regulated mechanism is mediated in part by post-translational modifications (PTM) of histones. These modifications dictate histone’s interactions with other proteins and modulate the reorganization of chromatin topology, two mechanisms which can modulate transcription. Indeed, some histone PTM exacerbate a repressive control over gene expression, while others promote it [70].
Many chromatin modifier enzymes are encoded by FMRP mRNA targets [71]. Reports of widespread dysregulation of histone PTM have been described in the mouse model of FXS, thus supporting this observation. Indeed, Korb et al. have monitored an increase of several histone modifications associated with an active chromatin topology in cortex neurons of Fmr1 KO mice [72]. Ensuing RNA-seq analysis consequently revealed more upregulated genes among all dysregulated transcripts. Furthermore, they showed that pharmacological inhibition of the transcription factor Brd4 (a protein that binds to acetylated histones and promotes transcription [73]) normalize spine morphology, aberrant behavior and partially rescue gene expression in the same model [72]. A more recent report has described an overall increase in H3 lysine 36 trimethylation in the hippocampus of the Fmr1 KO mouse [74], a PTM also associated with active gene expression [70].
Those studies highlighted the fact that the loss of FMRP promotes changes in chromatin topology which will result in a dysregulation of gene expression. Mutations in chromatin remodeling genes have also been repeatedly associated with neurodevelopmental disorders such as autism and ID [75,76,77]. Taken as a whole, those observations suggest that the alteration in histone PTM observed in the Fmr1 KO mouse may cause aberrant gene expression, which in turn, underlie key fragile X phenotypes.
3. Transcriptomic Alterations in Fragile X Syndrome
By its capacity to bind both RNA and proteins, FMRP acts as an adapter to facilitate the interaction between those two classes of biomolecules. This pivotal function allows FMRP to mediate various RNA processing mechanisms other than its traditional ability to regulate translation by ribosome stalling or eIF4E sequestration (formation of a protein complex comprising FMRP and eIF4E resulting in the inhibition of translation initiation) [20].
3.1. FMRP Mediates Micro RNA-Related Interference
MicroRNAs (miRNAs) are 19–24 nucleotides long non-coding RNAs involves in post-transcriptional regulation of approximately 30% of human genes [78]. Upon transcription, newly synthesized primary miRNAs are processed into a partially unpaired stem-loop precursor (pre-miRNA) by the nuclear ribonuclease Drosha. Pre-miRNAs are then exported through exportin 5 to the cytoplasm, where they are further processed by the ribonuclease Dicer into double-stranded RNA. The functional strand of this RNA duplex makes up the mature miRNA and is loaded into the RISC complex (RNA induced silencing complex) through its association with members of the Argonaute (AGO) protein family. From there, miRNA-containing RISC complex binds to complementary mRNA and either promotes its degradation or inhibits its translation [79,80].
FMRP is a well-established effector of the miRNA pathway. Indeed, FMRP is found in RISC and directly interacts with different components of those ribonucleoprotein (RNP) complexes, including AGO1, AGO2 and Dicer and binds to both mature and pre-miRNAs [81,82,83,84,85]. FMRP binding to miRNAs is mediated by its KH domains and is believed to favor miRNA annealing to their complementary mRNA [83]. As such, in vitro experiments have shown that the presence of FMRP in RISC complex can both promote and inhibit miRNAs-related interference. The function of FMRP in that miRNA-containing RNP complex is modulated by many factors, including the affinity of FMRP to the targeted mRNA and protein-protein interactions [81,83,86].
First evidence of the involvement of the miRNA pathway into fragile X physiopathology was brought by Jin et al. [82]. In this study, they showed that a downregulation of AGO1 alters FMRP-related translational control and promotes certain FXS phenotypes. Moreover, this work establishes FXS as the first neurologic disease associated with alteration in the miRNA pathway. Since then, an accumulation of experimental evidence has linked such deficit to the absence of FMRP and, consequently, to mechanisms underlying fragile X physiopathology. For example, the miR-125b mediated translational repression of NR2A (a sub-unit of the glutamate receptor NMDA) is FMRP-dependent and regulates spines morphogenesis [87]. Defects in axon guidance can also be linked to aberrant miRNA regulation in FXS. Indeed, Halevy et al. have used induced pluripotent stem cells derived from fragile X subjects’ fibroblasts to associate a downregulation of hsa-miR-382 to this neuronal alteration [88]. FMRP was also shown to modulate the miR-125a mediated interference of PSD-95, a protein involved in post-synaptic AMPA receptors endocytosis and consequently, in synaptic plasticity regulation [89,90]. PSD-95 and miR-125a are respectively found up and downregulated in synaptoneurosomes of Fmr1 KO mice. Even more interestingly, a recent miRNA profiling performed in two independent cohorts has identified an increase of miR-125a in urine of fragile X boys [91].
The miRNA pathway represents a third mechanism, with ribosome stalling and eIF4E sequestration, through which FMR exerts its regulatory effect on translation [20]. Correction of an improper miRNA interference therefore represents a potent therapeutic approach to rectify FXS biochemical alterations [79,92]. Furthermore, as shown by Putkonen et al. [91], circulatory miRNA may also constitute a great source of peripheral biomarkers that can be used as quantitative and objective tools by clinicians [93].
3.2. Alternative Splicing and FMRP
The first insight of FMRP involvement in alternative splicing came from the study of Didiot et al. [94] in which they showed that FMRP affects alternative splicing of its own mRNA. Indeed, they showed that FMRP bind its own mRNA trough two G-quartets located closely to a known alternative spliced site in exon 15. Consequently, they observed that both overexpression and silencing of FMRP leads to aberrant FMR1 splicing pattern [94]. Further evidence of FMRP implication into a more widespread regulation of alternative splicing comes from studies conducted in drosophila. Indeed, the knock-down of dFMRP leads to the deregulation of more than 100 splicing events [95]. Moreover, only a handful of dFMRP mRNA targets have seen their splicing pattern influenced by this genetic manipulation [96]. Those observations were recently transposed into the mouse model of fragile X. Indeed, Shah et al. [74] reported a reduction of approximately 30% of exon skipping event in Fmr1 KO mice hippocampus. Furthermore, many of the genes found with defective splicing are linked to neuronal transmission and autism [74]. FMRP is also known to interact with various proteins involved in pre-mRNA alternative splicing, one of which being the splicing factor RBM14 [97,98]. Together, FMRP and RBM14 modulate the splicing of Tau and Protrudin, two genes involved in neuronal cells differentiation and dendritic spines growth. The improper splicing pattern of those two genes observed in the hippocampus of Fmr1 KO mice therefore suggests that FMRP involvement in the regulation of pre-mRNA splicing can contribute to FXS physiopathology [98].
Alternative splicing is one of the most relevant mechanisms involved in the temporal and cell-specific regulation of gene expression and in the promotion of the proteome functional diversity. Maintenance of its homeostasis is thus mandatory to ensure appropriate cell function, especially in neural tissue where it participates in virtually all facets of neurons activity [99]. Despites mounting evidence regarding FMRP role in alternative splicing, the exact functions of FMRP in this molecular mechanism remain to be fully elucidated. More importantly, no study has yet corroborated those observations in specimens derived from fragile X patients.
3.3. FMRP Absence May Lead to Defect in RNA Editing
The ADAR (adenosine deaminase acting on RNA) family of enzymes catalyzes the post-transcriptional modification of adenosine to inosine in double-strand pre-mRNA. Inosine is interpreted as guanosine by the translational and splicing machinery. As such, A to I editing in mRNA may lead to variable amino acids incorporation into corresponding proteins and contribute to the regulation of its alternative splicing. In other words, RNA editing represents another mechanism by which different protein variants can emanate from one genomic entity. Furthermore, proper ADAR editing is believed to be crucial for normal neuronal functions, as many mRNA edited by those enzymes encode ion channels, neurotransmitter receptors and protein involved in synaptic transmission [100].
The initial observation of FMRP presence into ADAR-containing RNP complexes raised the possibility that FMRP may act has an RNA editing mediator [101]. Co-immunoprecipitation experiments report a direct interaction between FMRP and ADAR2, a partnership that was shown to inhibit ADAR2 RNA editing activity [102,103]. FMRP-dependent dysregulation of RNA editing was further observed in various models of FXS. Indeed, both FMRP silencing and mutations impairing KH domains RNA binding have proved to increase ADAR activity and alter editing for some of the mRNA studied [101,102,103]. These transcripts, which include the calcium channel Cav1.3 and glutamate receptor subunits GluK2, GluA2 and GluA4, are involved in synaptic modulation, thereby suggesting that RNA editing impairment may significantly contribute to FXS physiopathology [102,103]. However, those studies only considered the editing pattern of a preselected pool of mRNAs. A transcriptome wide characterization of RNA editing defects in fragile eventually clarify involvement of FMRP in RNA editing and the contribution of this mechanism to FXS phenotype.
4. Proteomics Alterations in Fragile X Syndrome
Expression patterns of numerous proteins are known to be deregulated in FXS. Those proteins exhibit a wide range of biological and cellular functions, ranging from receptors, enzymes, translation factors and more. The expected pathophysiological consequences of FMRP’s absence should therefore be as various as the nature of the proteins found improperly expressed in FXS.
4.1. Rate of Protein Synthesis Alteration: A Still Misunderstood Hallmark of FXS
Changes in the rate of protein synthesis remains a focal point of fragile X physiopathology given the general agreement regarding FMRP as a translational regulator. The first reported evidence of FMRP’s ability to regulate translation came from studies conducted in rabbit reticulocyte lysate, in which it was shown to inhibit the general translational rate in a dose-dependent manner [104,105]. Since then, a plethora of studies have employed a variety of FXS animal models, in combination with a profusion of experimental workflow, to firmly establish the relation between the absence of FMRP and an increased rate of protein synthesis [106,107,108].
Most of the work addressing this issue has been performed in order to conceptualize the mGluR theory, and consequently, in hippocampal slices prepared from Fmr1 KO mice. This experimental approach, which allows the combination of electrophysiological and metabolic measurements, has greatly contributed to the advancement of knowledge regarding synaptic and biochemical defects underlying the absence of FMRP and supports the development of various preclinical studies [43,109]. In fact, rescue of the fragile X phenotype in Fmr1 KO mice following targeted pharmacological treatments has repeatedly been associated with a normalization of the aberrant protein synthesis [44,45,47,110,111]. Such reports strengthen the role of FMRP in the maintenance of translational homeostasis to ensure normal neurologic and behavioral phenotypes and further establish the rate of protein synthesis alteration as one of the most potent monitoring biomarkers for FXS.
Despite being one of the most prominent and studied alterations in animal models, only a limited number of reports have studied protein synthesis defects in human, mainly due to limitations regarding sample nature and accessibility. Two studies have used fibroblasts obtained from a skin biopsy of FXS patients to monitor an increase in translational rate in fragile X derived cells [112,113]. Another substantial contribution concerning this issue came from two studies of the Beebe Smith group, in which positron emission tomography was used to measure a decrease of the in vivo integration of [11C] leucine into nascent brain proteins of 15 FXS patients under propofol sedation and a similar, but not significative, trend of perturbation in 9 FXS patients under dexmedetomidine anesthesia [114,115]. Finally, we recently reported that the rate of protein synthesis is also decreased in freshly extracted peripheral blood mononuclear cells (PBMCs) of FXS patients (Dionne et al., 2021, accepted, DOI: 10.1371/journal.pone.0251367). These studies undoubtedly shown that the perturbation in translational homeostasis found in animal models is replicated in fragile X patients. Furthermore, all these studies made the common observation that some FXS patients displayed a rate of protein synthesis within control range, thereby suggesting that any therapeutic intervention aimed at restoring translational homeostasis may not be beneficial for all of them. The tissue-specific trend of perturbation and variability observed into the FXS population seemingly demonstrates that the protein synthesis defects inherent to the absence of FMRP is more nuanced and complex in humans than in mice. Indeed, protein synthesis in KO mice was constantly shown to be solely upregulated in all tissue or cell types tested so far [113,116,117].
These observations clearly demonstrated that defects regarding protein synthesis are still misunderstood in humans, and that several questions remain to be addressed to clarify the situation. The first one would be to establish if normalization of the aberrant protein synthesis rate can be achieved by a targeted pharmacological treatment, and further confirm the usefulness of the rate of protein synthesis measurement as a monitoring biomarker for FXS. The development of such an objective and quantitative tool would represent a major step forward for any future FXS clinical trials [118]. Another substantial advancement would be to identify the proteins that are dysregulated during the timeframe that protein synthesis rate measurement is conducted. The same experimental approach could be used to study how FXS cells respond to different stimuli. Such experiments could provide valuable information on molecular mechanism underlying fragile X physiopathology and yield a relevant source of peripheral biomarkers [116].
4.2. Cell Signaling Defects in FXS
Cell signaling is a complex communication network that governs cell fundamental mechanisms and coordinate their activity. Through this process, extracellular signals activate specific cell surface receptors before being transduced by signaling cascades into a wide variety of intracellular responses. The Ras/Raf/MEK/ERK and the PI3K/AKT/mTOR pathways are two major actors of those cell signaling transduction mechanisms. Indeed, those ubiquitous pathways are pivotal for a broad range of cellular (cell proliferation and differentiation, transcription and translation regulation) and physiological (metabolism, synaptic plasticity, development) processes [119,120].
FMRP, through its ability to regulate the translation of its targeted mRNA, acts as a chief mediator of the two aforementioned pathways. Indeed, analyses conducted in different mouse brain regions and HEK293 cells have shown that FMRP binds the mRNA of various effectors and regulators of those signaling cascades [25,71,121]. Consequently, a basal hyperphosphorylation of ERK and AKT (reflecting the hyperactivation of their respective pathways) as well as a reduced ERK activation following Gp1 mGLUR stimulation were observed in the hippocampus and cortex of Fmr1 KO mice [122,123,124,125]. Those alterations can be directly linked to exaggerated mGluR5 signaling and contribute to the aberrant synaptic phenotype and dysregulated protein synthesis rate observed in this model. As such, the normalization of ERK and AKT phosphorylation status in KO mice have frequently been associated with the correction of the improper translational rate, LTD and behavioral phenotypes; thereby establishing measurements of ERK and AKT activation status as one of the most potent biomarkers of therapeutic efficiency for FXS [44,45,109,110,126].
The hyperactivation of the Ras/Raf/MEK/ERK and PI3K/AKT/mTOR pathways found in the brain of KO mice is also observable in a variety of human samples. Indeed, an increase in the phosphorylation of MEK, ERK, AKT and mTOR have been described in post-mortem brains of FX patients [127,128]. These molecular alterations are also replicated in more accessible and less invasive tissues, such as fibroblasts, PBMCs and blood platelets [112,127,129]. Most notably, our research group showed that the phosphorylation of ERK and AKT measured in blood platelets is correlated with the IQ of FXS patients [129]. Furthermore, a three-month lovastatin treatment corrected the exaggerated ERK overactivation in platelets and has been associated with a clinical improvement [130]. However, no study has yet shown that a reduction of the AKT pathway hyperactivation is achievable following a targeted treatment in human trials.
Taken together, these observations clearly illustrate the relevance of using cell signaling alterations in FXS as biomarkers of therapeutic efficiency in human trials. In addition, it could also be interesting to study whether the signaling defects observed outside of neurons can contribute to the physiopathology of FXS. Given the multifunctional nature of these signaling pathways and the fact that their hyperactivation is replicated in peripheral cells, it is very likely that their deregulation in non-neuronal tissues could contribute, to some extent, to a number of extra-neuronal manifestations or symptoms.
4.3. The Matrix Metalloproteinase-9 Involvement in FXS Physiopathology
The matrix metalloproteinase-9 (MMP9) is a zinc-dependent endopeptidase which targets many components of the extracellular matrix (ECM). In neuronal tissue, MMP9 enzymatic activity towards the perineuronal net (a network of loosely organized ECM components that supports several synaptic processes [131,132]) promotes dendritic spines elongation and regulates synaptic plasticity [133,134,135,136].
Colocalization studies conducted in mouse neurons have shown that MMP9 mRNA is bound by FMRP and subjected to its translational control [137]. Consequently, an increase in MMP9 expression has been reported in the frontal cortex and hippocampus of FXS patients and Fmr1 KO mice [137]. Furthermore, genetic deletion of MMP9 in Fmr1 KO mice rescued several FXS phenotypes, including the aberrant spine morphogenesis and synaptic plasticity [138]. Those observations prompted the uses of minocycline, an MMP9 inhibitor, as a targeted treatment for FXS [139].
Minocycline treatments were shown to successfully reduce MMP9 activity, reverse the improper spine morphogenesis and improve anxiolytic behavior and locomotor activity in Fmr1 KO mice [140,141]. During clinical trials, minocycline mildly improved FXS patient’s behavior, with a more pronounced effect on irritability and anxiety [142,143]. Dziembowska et al., further reported that the increased MMP9 plasma activity is reduced in some FXS patients following a three-month minocycline treatment [144]. Even though they did not observe any correlation between MMP9 inhibition and global clinical improvement, their results strongly suggest that plasma measurement of MMP9 can be used as a monitoring biomarker for FXS. A thorough validation within a larger FXS cohort is, however, mandatory to confirm that statement.
Those reports clearly demonstrated that MMP9 is a key contributor to FXS physiopathology. However, the precise mechanism by which MMP9 activity contributes to fragile X phenotype remains to be fully elucidated. Despites this, measurement of MMP9 proteolytic activity remains one of the promising biomarkers of FXS, as shown by the fact that the MMP9 dysregulation monitored in KO mice and post-mortem patient brains is replicated in FXS blood.
4.4. Aberrant Cytokines Profile: A Sign of Immune Dysfunction in FXS?
Cytokines are crucial mediators of cell signaling events occurring within the immune system and are secreted in response to various stimuli. A wide variety of extra-immune cells, including neurons and microglia, express cytokines and their respective receptors. Cytokines are consequently involved in the regulation of several neuronal functions, in both developing and mature brains, as well as in the nervous system response to infections and injuries. Such processes are important for normal brain operation, since they can modulate cognition and emotional processing [145].
Very little is known about a possible immune dysfunction in FXS since only a handful of exploratory studies have been reported. As such, an in situ comparative transcriptomic analysis has shown that dysregulated transcript from Fmr1 KO mouse embryos hippocampus and cortex primarily harbor an immunological signature [146]. A further study observed a decrease in IL-6 and TNF-α mRNA expression in Fmr1 KO mouse adult hippocampus [147]. Moreover, FXS PBMCs show an increase in the production of pro-inflammatory cytokines IL-6 and IL-12p40 after being conjointly stimulated with lipopolysaccharides (LPS) and DHPG, a Gp1 mGluR agonist. Interestingly, FXS leukocytes did not display any alteration in cytokine production when solely stimulated with LPS [148]. However, two studies have shown that circulating levels of several pro-inflammatory cytokines are actually decreased in FXS serum and plasma [149,150]. Despites being somewhat contradictory, those reports strongly suggest the existence of immune dysfunctions in FXS. This statement is supported by a recent systemic analysis of the medical diagnoses performed on 5736 FXS patients. Indeed, Yu et al. found a higher prevalence of various infectious diseases and an underrepresentation of autoimmune disorders in the FXS population [151].
An alteration in cytokines secretion or response could contribute to several FXS phenotypes, as it might increase the patient’s sensitivity to neurological damage induced by environmental, pathological or xenobiotic exposure. However, more detailed studies are needed to better characterize the potential immune dysfunctions found in FXS and the mechanism by which the absence of FMRP leads to such impairments.
5. Metabolomic Alterations in Fragile X Syndrome
Many studies have associated an aberrant metabolic profile with FMRP’s loss of expression. Indeed, metabolomic screenings conducted in Fmr1 KO mice and patients have shown that glucose, lipid, and several neurotransmitter metabolisms are deregulated in FXS [152,153]. The fact that FMRP binds to the mRNA of many key regulators of metabolism may explain those observations [71,152]. Above all, those results show that the extra-neuronal phenotype caused by FMRP’s absence must not be underestimated and, therefore, deserves to be better characterized.
5.1. The Hypocholesterolemic Phenotype of FXS
Cholesterol is a ubiquitous lipid and an essential component of eukaryotic membranes. It is known to play an important role in the maintenance of plasma membranes structural integrity and in the regulation of its functions [154]. Approximately 25% of all cholesterol within human body is found in the brain. Indeed, a large portion of this cholesterol is located in the myelin sheaths of axons and in dendritic spines membranes [155]. Maintenance of cholesterol homeostasis is therefore crucial for proper neurodevelopment and cognitive functions [156].
A prime example of a neurodevelopmental disease caused by an improper cholesterol metabolism is the Smith-Lemli-Opitz syndrome (SLOS), which exhibit mild to moderate ID and ASD as one of its most prevalent comorbidities [157,158]. SLOS is a recessive condition caused by mutations in the gene encoding the 7-dehydrocholesterol reductase, which catalyzes the last step of cholesterol biosynthesis. As such, SLOS patients typically present low levels of circulating cholesterol [158]. A higher prevalence of hypocholesterolemia was also found in an etiologically diverse autistic population [159,160,161]. Moreover, very low levels of circulating cholesterol (below the 10th centiles) were associated with higher risk of ID, anxiety and depression in men with ASD [161].
These observations promoted the investigation of peripheric levels of cholesterol in FXS. Indeed, two independent reports have observed lower plasma levels of LDL (light density lipoprotein), HDL (high-density lipoprotein) and total cholesterol in FXS men [162,163]. Several parameters of those cholesterol profiles were also shown to be correlated with the severity of the behavioral impairments found in these patients. Interestingly, no associations were observed between plasma level of the proprotein convertase subtilisin/kexin type 9 (PCSK9) and total/LDL cholesterol in FXS patients, despite the existence of a clear correlation in control individuals [163]. PCSK9 is secreted from hepatocytes and binds to LDL receptors, which promotes their degradation and favorite higher plasma concentration of cholesterol. Moreover, a recent report showed that the phosphorylation level of PCSK9, a PTM that enhances its binding affinity for the LDL receptor, is decreased in samples from FXS patients [164]. These observations, which suggest a dysregulation of PCSK9 activity in FXS, therefore provides the first clues regarding the molecular mechanism responsible for the hypocholesterolemia found in this population.
Since hypocholesterolemia is a common feature of FXS and ASD, it would be interesting to investigate if the same biochemical mechanisms underlying this phenotype are shared between both conditions. Such research could provide a better understanding of both diseases physiopathology and potentially pave the way for therapeutic approach that could be beneficial for individuals with FXS and ASD. Furthermore, it would also be interesting to validate if low cholesterol level could be used has prognostic marker of ASD and other comorbidities in FXS patients.
5.2. Cyclic AMP Metabolism Is Defective in Fragile X
Cyclic AMP (cAMP) is a second messenger involved in the transduction of a plethora of signaling pathways. The intracellular levels of cAMP are regulated by two classes of enzymes: adenylate cyclase (AC) and phosphodiesterase (PDE). Adenylate cyclase is responsible for cAMP synthesis. The majority (9 out of 10) of AC isoforms are membrane-bound enzymes that are regulated by various G protein-coupled receptors (GPCR) [165]. On the other hand, PDE catalyze cyclic nucleotides (cAMP and cGMP) degradation. Some PDE subfamilies are cAMP (PDE4,7 and 8) or cGMP (PDE5,6 and 9) specific, while the other (PDE1,2,3,10 and 11) act on both cyclic nucleotides [166]. Intracellular levels of cAMP promote the activity of various kinases such as PKA and ERK [167] and regulate gene expression through the activation of the transcription factor CREB [165]. As such, cAMP is involved in several cognitive and neuronal processes, including stress, anxiety, memory, synaptic plasticity, synaptic transmission and spine morphogenesis [168,169,170]
The pioneering work of Berry-Kravis et al. in the 1990s provided the first evidence of a defective cyclic AMP metabolism in FXS. Indeed, they reported that blood platelets and lymphoblastoid cell lines from FXS patients produced lower levels of cAMP compared to controls and other matched individuals with ID and ASD [171,172,173]. Furthermore, they reported that FMRP overexpression in mouse neurons enhance cAMP production, thereby highlighting the contribution of FMRP in cAMP pathway [174]. These observations were further replicated in animal models of FXS, more precisely the Fmr1 KO mouse and the dFMR1 null drosophila [175].
These studies paved the way for the elaboration of the “cAMP theory of fragile X”, which lays its foundation in the aberrant expression pattern of proteins involved in the cAMP cascade. Indeed, mRNAs of several AC isoforms and members of PDE subfamilies are targeted by FMRP [71,121]. This theory further states that the downregulation of cAMP signaling, both downstream and independently of Gp1 mGluR activation, contributes to FXS physiopathology [176]. This theory is corroborated by several studies conducted in KO mice, in which the reversal of several FXS phenotypes was associated with an upregulation of the cAMP pathway. Indeed, pharmacologic treatments that either stimulate adenylate cyclase activity, both directly or through the activation of AC-coupled GPCR or inhibit specific PDE have prove to be efficient therapeutic approach [177,178,179,180].
The accumulation of evidence regarding the defects in the cyclic AMP signaling in FXS and their contribution to the disease physiopathology are undeniable. As such, many effectors of this pathway represent potent therapeutic targets. Furthermore, as shown by the work of Berry-Kravis et al., quantification of cAMP production in peripheral blood cells could serve as objective biomarkers to assay treatment efficacy.
5.3. The Amyloid-β Precursor Protein and Its Secreted Metabolites
The Amyloid precursor protein (APP) is a dendritic transmembrane protein which is involved in several neuronal functions, including axonogenesis, neurite outgrowth and neuronal adhesion [181]. APP can be proteolytically processed by two distinct pathways. When undergoing the amyloidogenic pathway, APP is sequentially cleaved by the β-secretase BACE1 and the γ-secretase complex to produce the neurotoxic secreted β-APP (sAPPβ) and the amyloid-β (Aβ) peptides, which either takes the form of a 40 (Aβ40) or 42 (Aβ42) amino acids long chains. Aβ peptides are the major component of the amyloid plaques found in the brain of patients with Alzheimer’s disease and are therefore associated with neurodegeneration. Alternatively, APP can be processed by α-secretases to generate the secreted α-APP (sAPPα), which similarly to APP, bears a wide array of neurotrophic functions [182].
FMRP was shown to bind to APP mRNA and to inhibit its translation through interactions with the miRNA machinery [183]. As such, levels of APP, sAPPα and Aβ peptides were shown to be increased in brains of Fmr1 KO mice and linked to several key alterations composing the FXS phenotype [184,185]. In accordance with those observations, APP haploinsufficiency was associated with the rescue of repetitive behavior, anxiety, mGluR-LTD and spine morphology in Fmr1 KO mice [186]. Those alterations regarding APP expression and catabolism are also replicated in humans. Indeed, levels of APP, sAPPα and Aβ peptides were shown to be upregulated in post-mortem brains samples and various peripheral cells from FXS patients [184,186]. Plasma levels of both forms of secreted APP and Aβ peptides were also shown to be increased in children with FXS, while only Aβ42 seems to be decreased in adult patients. Interestingly, circulating levels of secreted APP and Aβ peptides were also shown to be deregulated in children with ASD. Indeed, samples collected from those patients showed an increased in sAPPα and a decreased in sAPPβ and both form of Aβ peptides [187,188,189,190]. Moreover, levels of sAPPα and total sAPP were shown to be reduced following a treatment with acamprosate in children with ASD, both with and without FXS [191].
The dysfunctions regarding APP expression and processing monitored in FXS suggest that those defects could contribute significantly to the mechanisms underlying the disease physiopathology. Furthermore, peripheral levels of APP and its secreted metabolites represent promising biomarkers for the monitoring of treatment efficacy and for the prediction of ASD comorbidities in FXS patients.
6. Conclusions
FMRP is a multifunctional protein that plays a role in several cellular and biochemical processes. The phenotypical consequences of its absence are diverse, and many are still misunderstood. Future research should focus on the elucidation of those unanswered questions. Such discovery would promote a better comprehension of the overall mechanism underlying FXS physiopathology and pave the way for new therapeutic approaches and the establishment of objective biomarkers to improve clinical management.
Author Contributions
Writing—original draft preparation, O.D.; writing—review and editing, F.C. and O.D.; visualization, O.D. Both authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Peprah, E. Fragile X Syndrome: The FMR1 CGG Repeat Distribution among World Populations. Ann. Hum. Genet. 2012, 76, 178–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, R.; Azarang, A.; Wilaisakditipakorn, T.; Hagerman, R.J. Fragile X Syndrome: A Review of Clinical Management. Intractable Rare Dis. Res. 2016, 5, 145–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciaccio, C.; Fontana, L.; Milani, D.; Tabano, S.; Miozzo, M.; Esposito, S. Fragile X Syndrome: A Review of Clinical and Molecular Diagnoses. Ital. J. Pediatr. 2017, 43. [Google Scholar] [CrossRef] [Green Version]
- Kidd, S.A.; Lachiewicz, A.; Barbouth, D.; Blitz, R.K.; Delahunty, C.; McBrien, D.; Visootsak, J.; Berry-Kravis, E. Fragile X Syndrome: A Review of Associated Medical Problems. Pediatrics 2014, 134, 995–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldarriaga, W.; Tassone, F.; González-Teshima, L.Y.; Forero-Forero, J.V.; Ayala-Zapata, S.; Hagerman, R. Fragile X Syndrome. Colomb Med. 2014, 45, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Verkerk, A.J.M.H.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.A.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a Gene (FMR-1) Containing a CGG Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Pieretti, M.; Warren, T.; Thomas, C.; Nelson’, D.L. Absence of Expression of the HIM-7 Gene in Fragile X Syndrome. Cell 1997, 66, 817–822. [Google Scholar] [CrossRef]
- Rousseau, F.; Heitz, D.; Tarleton, J.; MacPherson, J.; Malmgren, H.; Dahl, N.; Barnicoat, A.; Mathew, C.; Mornet, E.; Tejada, I. A Multicenter Study on Genotype-Phenotype Correlations in the Fragile X Syndrome, Using Direct Diagnosis with Probe StB12.3: The First 2,253 Cases. Am. J. Hum. Genet. 1994, 55, 225–237. [Google Scholar]
- Nolin, S.L.; Glicksman, A.; Houck, G.E.; Brown, W.T.; Dobkin, C.S. Mosaicism in Fragile X Affected Males. Am. J. Med. Genet. 1994, 51, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Pretto, D.; Yrigollen, C.M.; Tang, H.-T.; Williamson, J.; Espinal, G.; Iwahashi, C.K.; Durbin-Johnson, B.; Hagerman, R.J.; Hagerman, P.J.; Tassone, F. Clinical and Molecular Implications of Mosaicism in FMR1 Full Mutations. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Lessard, M.; Chouiali, A.; Drouin, R.; Sébire, G.; Corbin, F. Quantitative Measurement of FMRP in Blood Platelets as a New Screening Test for Fragile X Syndrome. Clin. Genet. 2012, 82, 472–477. [Google Scholar] [CrossRef]
- Willemsen, R. Predictive Testing for Cognitive Functioning in Female Carriers of the Fragile X Syndrome Using Hair Root Analysis. J. Med. Genet. 2003, 40, 377–379. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Hessl, D.; Randol, J.L.; Espinal, G.M.; Schneider, A.; Protic, D.; Aydin, E.Y.; Hagerman, R.J.; Hagerman, P.J. Association between IQ and FMR1 Protein (FMRP) across the Spectrum of CGG Repeat Expansions. PLoS ONE 2019, 14, e0226811. [Google Scholar] [CrossRef] [Green Version]
- Hinds, H.L.; Ashley, C.T.; Sutcliffe, J.S.; Nelson, D.L.; Warren, S.T.; Housman, D.E.; Schalling, M. Tissue Specific Expression of FMR–1 Provides Evidence for a Functional Role in Fragile X Syndrome. Nat. Genet. 1993, 3, 36–43. [Google Scholar] [CrossRef]
- Bakker, C.E.; de Diego Otero, Y.; Bontekoe, C.; Raghoe, P.; Luteijn, T.; Hoogeveen, A.T.; Oostra, B.A.; Willemsen, R. Immunocytochemical and Biochemical Characterization of FMRP, FXR1P, and FXR2P in the Mouse. Exp. Cell Res. 2000, 258, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Patzlaff, N.E.; Shen, M.; Zhao, X. Regulation of Adult Neurogenesis by the Fragile X Family of RNA Binding Proteins. Brain Plast. 2018, 3, 205–223. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.C.; Fraser, C.E.; Mostovetsky, O.; Stefani, G.; Jones, T.A.; Eddy, S.R.; Darnell, R.B. Kissing Complex RNAs Mediate Interaction between the Fragile-X Mental Retardation Protein KH2 Domain and Brain Polyribosomes. Genes Dev. 2005, 19, 903–918. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.C.; Jensen, K.B.; Jin, P.; Brown, V.; Warren, S.T.; Darnell, R.B. Fragile X Mental Retardation Protein Targets G Quartet MRNAs Important for Neuronal Function. Cell 2001, 107, 489–499. [Google Scholar] [CrossRef]
- Bechara, E.G.; Didiot, M.C.; Melko, M.; Davidovic, L.; Bensaid, M.; Martin, P.; Castets, M.; Pognonec, P.; Khandjian, E.W.; Moine, H.; et al. A Novel Function for Fragile X Mental Retardation Protein in Translational Activation. PLoS Biol. 2009, 7, e1000016. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.; Joseph, S. Fragile X Mental Retardation Protein: A Paradigm for Translational Control by RNA-Binding Proteins. Biochimie 2015, 114, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siomi, H.; Choi, M.; Siomi, M.C.; Nussbaum, R.L.; Dreyfuss, G. Essential Role for KH Domains in RNA Binding: Impaired RNA Binding by a Mutation in the KH Domain of FMR1 That Causes Fragile X Syndrome. Cell 1994, 77, 33–39. [Google Scholar] [CrossRef]
- Myrick, L.K.; Nakamoto-Kinoshita, M.; Lindor, N.M.; Kirmani, S.; Cheng, X.; Warren, S.T. Fragile X Syndrome Due to a Missense Mutation. Eur. J. Hum. Genet. 2014, 22, 1185–1189. [Google Scholar] [CrossRef] [Green Version]
- Corbin, F.; Bouillon, M.; Fortin, A.; Morin, S.; Rousseau, F.; Khandjian, E.W. The Fragile X Mental Retardation Protein Is Associated with Poly(A)+ MRNA in Actively Translating Polyribosomes. Hum. Mol. Genet. 1997, 6, 1465–1472. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.C.; Klann, E. The Translation of Translational Control by FMRP: Therapeutic Targets for FXS. Nat. Neurosci. 2013, 16, 1530–1536. [Google Scholar] [CrossRef] [Green Version]
- Maurin, T.; Bardoni, B. Fragile X Mental Retardation Protein: To Be or Not to Be a Translational Enhancer. Front. Mol. Biosci. 2018, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, G.M.; Shim, S.; Hong, D.S.; Garrett, A.S.; Quintin, E.-M.; Marzelli, M.J.; Patnaik, S.; Lightbody, A.A.; Reiss, A.L. Neuroanatomical Abnormalities in Fragile X Syndrome during the Adolescent and Young Adult Years. J. Psychiatr. Res. 2018, 107, 138–144. [Google Scholar] [CrossRef]
- Hallahan, B.P.; Craig, M.C.; Toal, F.; Daly, E.M.; Moore, C.J.; Ambikapathy, A.; Robertson, D.; Murphy, K.C.; Murphy, D.G.M. In Vivo Brain Anatomy of Adult Males with Fragile X Syndrome: An MRI Study. Neuroimage 2011, 54, 16–24. [Google Scholar] [CrossRef]
- Hoeft, F.; Carter, J.C.; Lightbody, A.A.; Cody Hazlett, H.; Piven, J.; Reiss, A.L. Region-Specific Alterations in Brain Development in One- to Three-Year-Old Boys with Fragile X Syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 9335–9339. [Google Scholar] [CrossRef] [Green Version]
- Lombroso, P.J. Genetics of Childhood Disorders: XLVIII. Learning and Memory, Part 1: Fragile X Syndrome Update. J. Am. Acad. Child. Adolesc. Psychiatry 2003, 42, 372–375. [Google Scholar] [CrossRef] [Green Version]
- He, C.X.; Portera-Cailliau, C. The Trouble with Spines in Fragile X Syndrome: Density, Maturity and Plasticity. Neuroscience 2013, 251, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Contractor, A.; Klyachko, V.A.; Portera-Cailliau, C. Altered Neuronal and Circuit Excitability in Fragile X Syndrome. Neuron 2015, 87, 699–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, J.R.; Bartley, A.F.; Hays, S.A.; Huber, K.M. Imbalance of Neocortical Excitation and Inhibition and Altered UP States Reflect Network Hyperexcitability in the Mouse Model of Fragile X Syndrome. J. Neurophysiol. 2008, 100, 2615–2626. [Google Scholar] [CrossRef] [PubMed]
- Morin-Parent, F.; Champigny, C.; Lacroix, A.; Corbin, F.; Lepage, J.-F. Hyperexcitability and Impaired Intracortical Inhibition in Patients with Fragile-X Syndrome. Transl. Psychiatry 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, M.S.; Auerbach, B.D.; Bear, M.F. Fragile X Mental Retardation Protein and Synaptic Plasticity. Mol. Brain 2013, 6, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, A.C.; Hagerman, R.J. Serotonin Dysregulation in Fragile X Syndrome: Implications for Treatment. Intractable Rare Dis. Res. 2014, 3, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Protic, D.; Salcedo-Arellano, M.J.; Dy, J.B.; Potter, L.A.; Hagerman, R.J. New Targeted Treatments for Fragile X Syndrome. Curr. Pediatr. Rev. 2019, 15, 251–258. [Google Scholar] [CrossRef]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The MGluR Theory of Fragile X Mental Retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef]
- Dölen, G.; Bear, M.F. Role for Metabotropic Glutamate Receptor 5 (MGluR5) in the Pathogenesis of Fragile X Syndrome: Pathogenesis of Fragile X Syndrome. J. Physiol. 2008, 586, 1503–1508. [Google Scholar] [CrossRef]
- Braat, S.; Kooy, R.F. Insights into GABAAergic System Deficits in Fragile X Syndrome Lead to Clinical Trials. Neuropharmacology 2015, 88, 48–54. [Google Scholar] [CrossRef]
- Van der Aa, N.; Kooy, R.F. GABAergic Abnormalities in the Fragile X Syndrome. Eur. J. Paediatr. Neurol. 2020, 24, 100–104. [Google Scholar] [CrossRef]
- Hagerman, R.; Lozano, R.; Hare, E. Modulation of the GABAergic Pathway for the Treatment of Fragile X Syndrome. Neuropsychiatr. Dis. Treat. 2014, 1769. [Google Scholar] [CrossRef] [Green Version]
- D’Hulst, C.; Heulens, I.; Brouwer, J.R.; Willemsen, R.; De Geest, N.; Reeve, S.P.; De Deyn, P.P.; Hassan, B.A.; Kooy, R.F. Expression of the GABAergic System in Animal Models for Fragile X Syndrome and Fragile X Associated Tremor/Ataxia Syndrome (FXTAS). Brain Res. 2009, 1253, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Dölen, G.; Osterweil, E.; Rao, B.S.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of Fragile X Syndrome in Mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalon, A.; Sidorov, M.; Ballard, T.M.; Ozmen, L.; Spooren, W.; Wettstein, J.G.; Jaeschke, G.; Bear, M.F.; Lindemann, L. Chronic Pharmacological MGlu5 Inhibition Corrects Fragile X in Adult Mice. Neuron 2012, 74, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Osterweil, E.K.; Chuang, S.-C.; Chubykin, A.A.; Sidorov, M.; Bianchi, R.; Wong, R.K.S.; Bear, M.F. Lovastatin Corrects Excess Protein Synthesis and Prevents Epileptogenesis in a Mouse Model of Fragile X Syndrome. Neuron 2013, 77, 243–250. [Google Scholar] [CrossRef] [Green Version]
- de Vrij, F.M.S.; Levenga, J.; van der Linde, H.C.; Koekkoek, S.K.; De Zeeuw, C.I.; Nelson, D.L.; Oostra, B.A.; Willemsen, R. Rescue of Behavioral Phenotype and Neuronal Protrusion Morphology in Fmr1 KO Mice. Neurobiol. Dis. 2008, 31, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Henderson, C.; Wijetunge, L.; Kinoshita, M.N.; Shumway, M.; Hammond, R.S.; Postma, F.R.; Brynczka, C.; Rush, R.; Thomas, A.; Paylor, R.; et al. Reversal of Disease-Related Pathologies in the Fragile X Mouse Model by Selective Activation of GABAB Receptors with Arbaclofen. Sci. Transl. Med. 2012, 4, 152ra128. [Google Scholar] [CrossRef]
- Heulens, I.; D’Hulst, C.; Van Dam, D.; De Deyn, P.P.; Kooy, R.F. Pharmacological Treatment of Fragile X Syndrome with GABAergic Drugs in a Knockout Mouse Model. Behav. Brain Res. 2012, 229, 244–249. [Google Scholar] [CrossRef]
- Olmos-Serrano, J.L.; Corbin, J.G.; Burns, M.P. The GABA(A) Receptor Agonist THIP Ameliorates Specific Behavioral Deficits in the Mouse Model of Fragile X Syndrome. Dev. Neurosci. 2011, 33, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Qin, M.; Huang, T.; Kader, M.; Krych, L.; Xia, Z.; Burlin, T.; Zeidler, Z.; Zhao, T.; Smith, C.B. R-Baclofen Reverses a Social Behavior Deficit and Elevated Protein Synthesis in a Mouse Model of Fragile X Syndrome. Int. J. Neuropsychopharmacol. 2015, 18, pyv034. [Google Scholar] [CrossRef] [Green Version]
- Berry-Kravis, E.; Hessl, D.; Coffey, S.; Hervey, C.; Schneider, A.; Yuhas, J.; Hutchison, J.; Snape, M.; Tranfaglia, M.; Nguyen, D.V.; et al. A Pilot Open Label, Single Dose Trial of Fenobam in Adults with Fragile X Syndrome. J. Med. Genet. 2009, 46, 266–271. [Google Scholar] [CrossRef] [Green Version]
- Youssef, E.A.; Berry-Kravis, E.; Czech, C.; Hagerman, R.J.; Hessl, D.; Wong, C.Y.; Rabbia, M.; Deptula, D.; John, A.; Kinch, R.; et al. Effect of the MGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacology 2018, 43, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Hagerman, R.; Jacquemont, S.; Berry-Kravis, E.; Des Portes, V.; Stanfield, A.; Koumaras, B.; Rosenkranz, G.; Murgia, A.; Wolf, C.; Apostol, G.; et al. Mavoglurant in Fragile X Syndrome: Results of Two Open-Label, Extension Trials in Adults and Adolescents. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Jacquemont, S.; Curie, A.; des Portes, V.; Torrioli, M.G.; Berry-Kravis, E.; Hagerman, R.J.; Ramos, F.J.; Cornish, K.; He, Y.; Paulding, C.; et al. Epigenetic Modification of the FMR1 Gene in Fragile X Syndrome Is Associated with Differential Response to the MGluR5 Antagonist AFQ056. Sci. Transl. Med. 2011, 3, 64ra1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, C.A.; Weng, N.; Weiler, I.J.; Greenough, W.T.; Stigler, K.A.; Wink, L.K.; McDougle, C.J. Open-Label Riluzole in Fragile X Syndrome. Brain Res. 2011, 1380, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Ligsay, A.; Van Dijck, A.; Nguyen, D.V.; Lozano, R.; Chen, Y.; Bickel, E.S.; Hessl, D.; Schneider, A.; Angkustsiri, K.; Tassone, F.; et al. A Randomized Double-Blind, Placebo-Controlled Trial of Ganaxolone in Children and Adolescents with Fragile X Syndrome. J. Neurodev. Disord. 2017, 9, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry-Kravis, E.M.; Hessl, D.; Rathmell, B.; Zarevics, P.; Cherubini, M.; Walton-Bowen, K.; Mu, Y.; Nguyen, D.V.; Gonzalez-Heydrich, J.; Wang, P.P.; et al. Effects of STX209 (Arbaclofen) on Neurobehavioral Function in Children and Adults with Fragile X Syndrome: A Randomized, Controlled, Phase 2 Trial. Sci. Transl. Med. 2012, 4, 152ra127. [Google Scholar] [CrossRef] [Green Version]
- Berry-Kravis, E.; Hagerman, R.; Visootsak, J.; Budimirovic, D.; Kaufmann, W.E.; Cherubini, M.; Zarevics, P.; Walton-Bowen, K.; Wang, P.; Bear, M.F.; et al. Arbaclofen in Fragile X Syndrome: Results of Phase 3 Trials. J. Neurodev. Disord. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Erickson, C.A.; Wink, L.K.; Ray, B.; Early, M.C.; Stiegelmeyer, E.; Mathieu-Frasier, L.; Patrick, V.; Lahiri, D.K.; McDougle, C.J. Impact of Acamprosate on Behavior and Brain-Derived Neurotrophic Factor: An Open-Label Study in Youth with Fragile X Syndrome. Psychopharmacology 2013, 228, 75–84. [Google Scholar] [CrossRef]
- Dahlhaus, R. Of Men and Mice: Modeling the Fragile X Syndrome. Front. Mol. Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef]
- Zhao, X.; Bhattacharyya, A. Human Models Are Needed for Studying Human Neurodevelopmental Disorders. Am. J. Hum. Genet. 2018, 103, 829–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sittler, A.; Devys, D.; Weber, C.; Mandel, J.L. Alternative Splicing of Exon 14 Determines Nuclear or Cytoplasmic Localisation of Fmr1 Protein Isoforms. Hum. Mol. Genet. 1996, 5, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Gutekunst, C.-A.; Eberhart, D.E.; Yi, H.; Warren, S.T.; Hersch, S.M. Fragile X Mental Retardation Protein: Nucleocytoplasmic Shuttling and Association with Somatodendritic Ribosomes. J. Neurosci. 1997, 17, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Dury, A.Y.; Fatimy, R.E.; Tremblay, S.; Rose, T.M.; Côté, J.; Koninck, P.D.; Khandjian, E.W. Nuclear Fragile X Mental Retardation Protein Is Localized to Cajal Bodies. PLoS Genet. 2013, 9, e1003890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpatov, R.; Lesch, B.J.; Nakamoto-Kinoshita, M.; Blanco, A.; Chen, S.; Stützer, A.; Armache, K.J.; Simon, M.D.; Xu, C.; Ali, M.; et al. A Chromatin-Dependent Role of the Fragile X Mental Retardation Protein FMRP in the DNA Damage Response. Cell 2014, 157, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.J.; Yang, L.-X. Gamma-H2AX—A Novel Biomarker for DNA Double-Strand Breaks. In Vivo 2008, 22, 305–309. [Google Scholar]
- Bozzetti, M.P.; Specchia, V.; Cattenoz, P.B.; Laneve, P.; Geusa, A.; Sahin, H.B.; Di Tommaso, S.; Friscini, A.; Massari, S.; Diebold, C.; et al. The Drosophila Fragile X Mental Retardation Protein Participates in the PiRNA Pathway. J. Cell. Sci. 2015, 128, 2070–2084. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Lu, F.; Li, P.; Liu, W.; Zhao, L.; Wang, Q.; Cao, X.; Zhang, L.; Zhang, Y.Q. Drosophila Homolog of FMRP Maintains Genome Integrity by Interacting with Piwi. J. Genet. Genom. 2016, 43, 11–24. [Google Scholar] [CrossRef]
- Tóth, K.F.; Pezic, D.; Stuwe, E.; Webster, A. The PiRNA Pathway Guards the Germline Genome Against Transposable Elements. Adv. Exp. Med. Biol. 2016, 886, 51–77. [Google Scholar] [CrossRef] [Green Version]
- Fyodorov, D.V.; Zhou, B.-R.; Skoultchi, A.I.; Bai, Y. Emerging Roles of Linker Histones in Regulating Chromatin Structure and Function. Nat. Rev. Mol. Cell Biol. 2018, 19, 192–206. [Google Scholar] [CrossRef]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP Stalls Ribosomal Translocation on MRNAs Linked to Synaptic Function and Autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Korb, E.; Herre, M.; Zucker-Scharff, I.; Gresack, J.; Allis, C.D.; Darnell, R.B. Excess Translation of Epigenetic Regulators Contributes to Fragile X Syndrome and Is Alleviated by Brd4 Inhibition. Cell 2017, 170, 1209–1223.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond Transcriptional Regulation. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Molinaro, G.; Liu, B.; Wang, R.; Huber, K.M.; Richter, J.D. FMRP Control of Ribosome Translocation Promotes Chromatin Modifications and Alternative Splicing of Neuronal Genes Linked to Autism. Cell Rep. 2020, 30, 4459–4472.e6. [Google Scholar] [CrossRef]
- He, Q.; Ge, W. FMRP: A New Chapter with Chromatin. Protein Cell 2014, 5, 885–888. [Google Scholar] [CrossRef] [Green Version]
- Goodman, J.V.; Bonni, A. Regulation of Neuronal Connectivity in the Mammalian Brain by Chromatin Remodeling. Curr. Opin. Neurobiol. 2019, 59, 59–68. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, N.; Zhang, Y.; Du, Y.; Zhang, T.; Li, Z.; Wu, J.; Wang, X. Prioritized High-Confidence Risk Genes for Intellectual Disability Reveal Molecular Convergence During Brain Development. Front. Genet. 2018, 9, 349. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates That Thousands of Human Genes Are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Cao, T.; Zhen, X. Dysregulation of MiRNA and Its Potential Therapeutic Application in Schizophrenia. CNS Neurosci. Ther. 2018, 24, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Vishnoi, A.; Rani, S. MiRNA Biogenesis and Regulation of Diseases: An Overview. In MicroRNA Profiling: Methods and Protocols; Rani, S., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 1–10. ISBN 978-1-4939-6524-3. [Google Scholar]
- Caudy, A.A.; Myers, M.; Hannon, G.J.; Hammond, S.M. Fragile X-Related Protein and VIG Associate with the RNA Interference Machinery. Genes Dev. 2002, 16, 2491–2496. [Google Scholar] [CrossRef] [Green Version]
- Jin, P.; Zarnescu, D.C.; Ceman, S.; Nakamoto, M.; Mowrey, J.; Jongens, T.A.; Nelson, D.L.; Moses, K.; Warren, S.T. Biochemical and Genetic Interaction between the Fragile X Mental Retardation Protein and the MicroRNA Pathway. Nat. Neurosci. 2004, 7, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Plante, I.; Davidovic, L.; Ouellet, D.L.; Gobeil, L.-A.; Tremblay, S.; Khandjian, E.W.; Provost, P. Dicer-Derived MicroRNAs Are Utilized by the Fragile X Mental Retardation Protein for Assembly on Target RNAs. J. Biomed. Biotechnol. 2006, 2006, 64347. [Google Scholar] [CrossRef]
- Cheever, A.; Ceman, S. Phosphorylation of FMRP Inhibits Association with Dicer. RNA 2009, 15, 362–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, K.; Ishizuka, A.; Siomi, H.; Siomi, M.C. Distinct Roles for Argonaute Proteins in Small RNA-Directed RNA Cleavage Pathways. Genes Dev. 2004, 18, 1655–1666. [Google Scholar] [CrossRef] [Green Version]
- Kenny, P.J.; Zhou, H.; Kim, M.; Skariah, G.; Khetani, R.S.; Drnevich, J.; Arcila, M.L.; Kosik, K.S.; Ceman, S. MOV10 and FMRP Regulate AGO2 Association with MicroRNA Recognition Elements. Cell Rep. 2014, 9, 1729–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.-F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of Synaptic Structure and Function by FMRP-Associated MicroRNAs MiR-125b and MiR-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Halevy, T.; Czech, C.; Benvenisty, N. Molecular Mechanisms Regulating the Defects in Fragile X Syndrome Neurons Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2015, 4, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Muddashetty, R.S.; Nalavadi, V.C.; Gross, C.; Yao, X.; Xing, L.; Laur, O.; Warren, S.T.; Bassell, G.J. Reversible Inhibition of PSD-95 MRNA Translation by MiR-125a, FMRP Phosphorylation, and MGluR Signaling. Mol. Cell 2011, 42, 673–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMarco, B.; Stefanovic, S.; Williams, A.; Moss, K.R.; Anderson, B.R.; Bassell, G.J.; Mihailescu, M.R. FMRP—G-Quadruplex MRNA—MiR-125a Interactions: Implications for MiR-125a Mediated Translation Regulation of PSD-95 MRNA. PLoS ONE 2019, 14, e0217275. [Google Scholar] [CrossRef] [PubMed]
- Putkonen, N.; Laiho, A.; Ethell, D.; Pursiheimo, J.; Anttonen, A.-K.; Pitkonen, J.; Gentile, A.M.; de Diego-Otero, Y.; Castrén, M.L. Urine MicroRNA Profiling Displays MiR-125a Dysregulation in Children with Fragile X Syndrome. Cells 2020, 9, 289. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, B.C.; Ooi, J.Y.; Lin, R.C.; McMullen, J.R. MiRNA Therapeutics: A New Class of Drugs with Potential Therapeutic Applications in the Heart. Future Med. Chem. 2015, 7, 1771–1792. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.; Meese, E.; Keller, A. Specific MiRNA Disease Biomarkers in Blood, Serum and Plasma: Challenges and Prospects. Mol. Diagn. Ther. 2016, 20, 509–518. [Google Scholar] [CrossRef]
- Didiot, M.-C.; Tian, Z.; Schaeffer, C.; Subramanian, M.; Mandel, J.-L.; Moine, H. The G-Quartet Containing FMRP Binding Site in FMR1 MRNA Is a Potent Exonic Splicing Enhancer. Nucleic Acids Res. 2008, 36, 4902–4912. [Google Scholar] [CrossRef] [Green Version]
- Brooks, A.N.; Duff, M.O.; May, G.; Yang, L.; Bolisetty, M.; Landolin, J.; Wan, K.; Sandler, J.; Booth, B.W.; Celniker, S.E.; et al. Regulation of Alternative Splicing in Drosophila by 56 RNA Binding Proteins. Genome Res. 2015, 25, 1771–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoiber, M.H.; Olson, S.; May, G.E.; Duff, M.O.; Manent, J.; Obar, R.; Guruharsha, K.G.; Bickel, P.J.; Artavanis-Tsakonas, S.; Brown, J.B.; et al. Extensive Cross-Regulation of Post-Transcriptional Regulatory Networks in Drosophila. Genome Res. 2015, 25, 1692–1702. [Google Scholar] [CrossRef] [Green Version]
- Pasciuto, E.; Bagni, C. SnapShot: FMRP Interacting Proteins. Cell 2014, 159, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.-T.; Ye, S.-H.; Yang, H.-X.; Zhou, Y.-T.; Zhao, Q.-H.; Sun, W.-W.; Gao, M.-M.; Yi, Y.-H.; Long, Y.-S. A Novel Role of Fragile X Mental Retardation Protein in Pre-MRNA Alternative Splicing through RNA-Binding Protein 14. Neuroscience 2017, 349, 64–75. [Google Scholar] [CrossRef]
- Raj, B.; Blencowe, B.J. Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 2015, 87, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Krestel, H.; Meier, J.C. RNA Editing and Retrotransposons in Neurology. Front. Mol. Neurosci. 2018, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, B.; Jepson, J.E.; Savva, Y.A.; Pepper, A.S.-R.; Reenan, R.A.; Jongens, T.A. Modulation of DADAR-Dependent RNA Editing by the Drosophila Fragile X Mental Retardation Protein. Nat. Neurosci. 2011, 14, 1517–1524. [Google Scholar] [CrossRef]
- Shamay-Ramot, A.; Khermesh, K.; Porath, H.T.; Barak, M.; Pinto, Y.; Wachtel, C.; Zilberberg, A.; Lerer-Goldshtein, T.; Efroni, S.; Levanon, E.Y.; et al. Fmrp Interacts with Adar and Regulates RNA Editing, Synaptic Density and Locomotor Activity in Zebrafish. PLoS Genet. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Filippini, A.; Bonini, D.; Lacoux, C.; Pacini, L.; Zingariello, M.; Sancillo, L.; Bosisio, D.; Salvi, V.; Mingardi, J.; La Via, L.; et al. Absence of the Fragile X Mental Retardation Protein Results in Defects of RNA Editing of Neuronal MRNAs in Mouse. RNA Biol. 2017, 14, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Laggerbauer, B.; Ostareck, D.; Keidel, E.M.; Ostareck-Lederer, A.; Fischer, U. Evidence That Fragile X Mental Retardation Protein Is a Negative Regulator of Translation. Hum. Mol. Genet. 2001, 10, 329–338. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Ku, L.; Wilkinson, K.D.; Warren, S.T.; Feng, Y. The Fragile X Mental Retardation Protein Inhibits Translation via Interacting with MRNA. Nucleic Acids Res. 2001, 29, 2276–2283. [Google Scholar] [CrossRef] [Green Version]
- Bolduc, F.V.; Bell, K.; Cox, H.; Broadie, K.S.; Tully, T. Excess Protein Synthesis in Drosophila Fragile X Mutants Impairs Long-Term Memory. Nat. Neurosci. 2008, 11, 1143–1145. [Google Scholar] [CrossRef]
- Qin, M.; Kang, J.; Burlin, T.V.; Jiang, C.; Smith, C.B. Postadolescent Changes in Regional Cerebral Protein Synthesis: An in Vivo Study in the FMR1 Null Mouse. J. Neurosci. 2005, 25, 5087–5095. [Google Scholar] [CrossRef] [Green Version]
- Till, S.M.; Asiminas, A.; Jackson, A.D.; Katsanevaki, D.; Barnes, S.A.; Osterweil, E.K.; Bear, M.F.; Chattarji, S.; Wood, E.R.; Wyllie, D.J.A.; et al. Conserved Hippocampal Cellular Pathophysiology but Distinct Behavioural Deficits in a New Rat Model of FXS. Hum. Mol. Genet. 2015, 24, 5977–5984. [Google Scholar] [CrossRef] [Green Version]
- Osterweil, E.K.; Krueger, D.D.; Reinhold, K.; Bear, M.F. Hypersensitivity to MGluR5 and ERK1/2 Leads to Excessive Protein Synthesis in the Hippocampus of a Mouse Model of Fragile X Syndrome. J. Neurosci. 2010, 30, 15616–15627. [Google Scholar] [CrossRef] [Green Version]
- Gantois, I.; Khoutorsky, A.; Popic, J.; Aguilar-Valles, A.; Freemantle, E.; Cao, R.; Sharma, V.; Pooters, T.; Nagpal, A.; Skalecka, A.; et al. Metformin Ameliorates Core Deficits in a Mouse Model of Fragile X Syndrome. Nat. Med. 2017, 23, 674–677. [Google Scholar] [CrossRef] [PubMed]
- McCamphill, P.K.; Stoppel, L.J.; Senter, R.K.; Lewis, M.C.; Heynen, A.J.; Stoppel, D.C.; Sridhar, V.; Collins, K.A.; Shi, X.; Pan, J.Q.; et al. Selective Inhibition of Glycogen Synthase Kinase 3α Corrects Pathophysiology in a Mouse Model of Fragile X Syndrome. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Kumari, D.; Bhattacharya, A.; Nadel, J.; Moulton, K.; Zeak, N.M.; Glicksman, A.; Dobkin, C.; Brick, D.J.; Schwartz, P.H.; Smith, C.B.; et al. Identification of Fragile X Syndrome Specific Molecular Markers in Human Fibroblasts: A Useful Model to Test the Efficacy of Therapeutic Drugs. Hum. Mutat. 2014, 35, 1485–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemont, S.; Pacini, L.; Jønch, A.E.; Cencelli, G.; Rozenberg, I.; He, Y.; D’Andrea, L.; Pedini, G.; Eldeeb, M.; Willemsen, R.; et al. Protein Synthesis Levels Are Increased in a Subset of Individuals with Fragile X Syndrome. Hum. Mol. Genet. 2018, 27, 2039–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, M.; Schmidt, K.C.; Zametkin, A.J.; Bishu, S.; Horowitz, L.M.; Burlin, T.V.; Xia, Z.; Huang, T.; Quezado, Z.M.; Smith, C.B. Altered Cerebral Protein Synthesis in Fragile X Syndrome: Studies in Human Subjects and Knockout Mice. J. Cereb. Blood Flow Metab. 2013, 33, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, K.C.; Loutaev, I.; Quezado, Z.; Sheeler, C.; Smith, C.B. Regional Rates of Brain Protein Synthesis Are Unaltered in Dexmedetomidine Sedated Young Men with Fragile X Syndrome: A L-[1-11C]Leucine PET Study. Neurobiol. Dis. 2020, 143, 104978. [Google Scholar] [CrossRef] [PubMed]
- Bowling, H.; Bhattacharya, A.; Zhang, G.; Alam, D.; Lebowitz, J.Z.; Bohm-Levine, N.; Lin, D.; Singha, P.; Mamcarz, M.; Puckett, R.; et al. Altered Steady State and Activity-Dependent de Novo Protein Expression in Fragile X Syndrome. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Gross, C.; Nakamoto, M.; Yao, X.; Chan, C.-B.; Yim, S.Y.; Ye, K.; Warren, S.T.; Bassell, G.J. Excess Phosphoinositide 3-Kinase Subunit Synthesis and Activity as a Novel Therapeutic Target in Fragile X Syndrome. J. Neurosci. 2010, 30, 10624–10638. [Google Scholar] [CrossRef] [Green Version]
- Berry-Kravis, E.; Hessl, D.; Abbeduto, L.; Reiss, A.L.; Beckel-Mitchener, A.; Urv, T.K. Outcome Measures for Clinical Trials in Fragile X Syndrome. J. Dev. Behav. Pediatr. 2013, 34, 508–522. [Google Scholar] [CrossRef] [Green Version]
- Fayard, E. Protein Kinase B/Akt at a Glance. J. Cell Sci. 2005, 118, 5675–5678. [Google Scholar] [CrossRef] [Green Version]
- Buscà, R.; Pouysségur, J.; Lenormand, P. ERK1 and ERK2 Map Kinases: Specific Roles or Functional Redundancy? Front. Cell Dev. Biol. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Ascano, M.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. FMRP Targets Distinct MRNA Sequence Elements to Regulate Protein Expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef]
- Hou, L.; Antion, M.D.; Hu, D.; Spencer, C.M.; Paylor, R.; Klann, E. Dynamic Translational and Proteasomal Regulation of Fragile X Mental Retardation Protein Controls MGluR-Dependent Long-Term Depression. Neuron 2006, 51, 441–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, T.J.; Rashid, M.H.; Millecamps, M.; Sanoja, R.; Entrena, J.M.; Cervero, F. Decreased Nociceptive Sensitization in Mice Lacking the Fragile X Mental Retardation Protein: Role of MGluR1/5 and MTOR. J. Neurosci. 2007, 27, 13958–13967. [Google Scholar] [CrossRef]
- Weng, N.; Weiler, I.J.; Sumis, A.; Berry-Kravis, E.; Greenough, W.T. Early-Phase ERK Activation as a Biomarker for Metabolic Status in Fragile X Syndrome. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147B, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Hoeffer, C.A.; Takayasu, Y.; Miyawaki, T.; McBride, S.M.; Klann, E.; Zukin, R.S. Dysregulation of MTOR Signaling in Fragile X Syndrome. J. Neurosci. 2010, 30, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Kaphzan, H.; Alvarez-Dieppa, A.C.; Murphy, J.P.; Pierre, P.; Klann, E. Genetic Removal of P70 S6 Kinase 1 Corrects Molecular, Synaptic, and Behavioral Phenotypes in Fragile X Syndrome Mice. Neuron 2012, 76, 325–337. [Google Scholar] [CrossRef] [Green Version]
- Hoeffer, C.A.; Sanchez, E.; Hagerman, R.J.; Mu, Y.; Nguyen, D.V.; Wong, H.; Whelan, A.M.; Zukin, R.S.; Klann, E.; Tassone, F. Altered MTOR Signaling and Enhanced CYFIP2 Expression Levels in Subjects with Fragile X Syndrome. Genes Brain Behav. 2012, 11, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Snape, M.; Klann, E.; Stone, J.G.; Singh, A.; Petersen, R.B.; Castellani, R.J.; Casadesus, G.; Smith, M.A.; Zhu, X. Activation of the Extracellular Signal-Regulated Kinase Pathway Contributes to the Behavioral Deficit of Fragile x-Syndrome. J. Neurochem. 2012, 121, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, D.; Çaku, A.; Fradet, M.; Bouvier, P.; Dubé, J.; Corbin, F. Lovastatin Corrects ERK Pathway Hyperactivation in Fragile X Syndrome: Potential of Platelet’s Signaling Cascades as New Outcome Measures in Clinical Trials. Biomarkers 2016, 21, 497–508. [Google Scholar] [CrossRef]
- Çaku, A.; Pellerin, D.; Bouvier, P.; Riou, E.; Corbin, F. Effect of Lovastatin on Behavior in Children and Adults with Fragile X Syndrome: An Open-Label Study. Am. J. Med. Genet. Part A 2014, 164, 2834–2842. [Google Scholar] [CrossRef]
- Wlodarczyk, J.; Mukhina, I.; Kaczmarek, L.; Dityatev, A. Extracellular Matrix Molecules, Their Receptors, and Secreted Proteases in Synaptic Plasticity. Dev. Neurobiol. 2011, 71, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Dansie, L.E.; Ethell, I.M. Casting a Net on Dendritic Spines: The Extracellular Matrix and Its Receptors. Dev. Neurobiol. 2011, 71, 956–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaluk, P.; Wawrzyniak, M.; Alot, P.; Szczot, M.; Wyrembek, P.; Mercik, K.; Medvedev, N.; Wilczek, E.; De Roo, M.; Zuschratter, W.; et al. Influence of Matrix Metalloproteinase MMP-9 on Dendritic Spine Morphology. J. Cell Sci. 2011, 124, 3369–3380. [Google Scholar] [CrossRef] [Green Version]
- Reinhard, S.M.; Razak, K.; Ethell, I.M. A Delicate Balance: Role of MMP-9 in Brain Development and Pathophysiology of Neurodevelopmental Disorders. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [Green Version]
- Nagy, V.; Bozdagi, O.; Matynia, A.; Balcerzyk, M.; Okulski, P.; Dzwonek, J.; Costa, R.M.; Silva, A.J.; Kaczmarek, L.; Huntley, G.W. Matrix Metalloproteinase-9 Is Required for Hippocampal Late-Phase Long-Term Potentiation and Memory. J. Neurosci. 2006, 26, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bozdagi, O.; Nikitczuk, J.S.; Zhai, Z.W.; Zhou, Q.; Huntley, G.W. Extracellular Proteolysis by Matrix Metalloproteinase-9 Drives Dendritic Spine Enlargement and Long-Term Potentiation Coordinately. Proc. Natl. Acad. Sci. USA 2008, 105, 19520–19525. [Google Scholar] [CrossRef] [Green Version]
- Janusz, A.; Milek, J.; Perycz, M.; Pacini, L.; Bagni, C.; Kaczmarek, L.; Dziembowska, M. The Fragile X Mental Retardation Protein Regulates Matrix Metalloproteinase 9 MRNA at Synapses. J. Neurosci. 2013, 33, 18234–18241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhu, H.; Dansie, L.E.; Hickmott, P.W.; Ethell, D.W.; Ethell, I.M. Genetic Removal of Matrix Metalloproteinase 9 Rescues the Symptoms of Fragile X Syndrome in a Mouse Model. J. Neurosci. 2014, 34, 9867–9879. [Google Scholar] [CrossRef] [Green Version]
- Vandooren, J.; Knoops, S.; Aldinucci Buzzo, J.L.; Boon, L.; Martens, E.; Opdenakker, G.; Kolaczkowska, E. Differential Inhibition of Activity, Activation and Gene Expression of MMP-9 in THP-1 Cells by Azithromycin and Minocycline versus Bortezomib: A Comparative Study. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Dansie, L.E.; Phommahaxay, K.; Okusanya, A.G.; Uwadia, J.; Huang, M.; Rotschafer, S.E.; Razak, K.A.; Ethell, D.W.; Ethell, I.M. Long-Lasting Effects of Minocycline on Behavior in Young but Not Adult Fragile X Mice. Neuroscience 2013, 246, 186–198. [Google Scholar] [CrossRef] [Green Version]
- Bilousova, T.V.; Dansie, L.; Ngo, M.; Aye, J.; Charles, J.R.; Ethell, D.W.; Ethell, I.M. Minocycline Promotes Dendritic Spine Maturation and Improves Behavioural Performance in the Fragile X Mouse Model. J. Med. Genet. 2009, 46, 94–102. [Google Scholar] [CrossRef]
- Leigh, M.J.S.; Nguyen, D.V.; Mu, Y.; Winarni, T.I.; Schneider, A.; Chechi, T.; Polussa, J.; Doucet, P.; Tassone, F.; Rivera, S.M.; et al. A Randomized Double-Blind, Placebo-Controlled Trial of Minocycline in Children and Adolescents with Fragile x Syndrome. J. Dev. Behav. Pediatr. 2013, 34, 147–155. [Google Scholar] [CrossRef]
- Paribello, C.; Tao, L.; Folino, A.; Berry-Kravis, E.; Tranfaglia, M.; Ethell, I.M.; Ethell, D.W. Open-Label Add-on Treatment Trial of Minocycline in Fragile X Syndrome. BMC Neurol. 2010, 10, 91. [Google Scholar] [CrossRef] [Green Version]
- Dziembowska, M.; Pretto, D.I.; Janusz, A.; Kaczmarek, L.; Leigh, M.J.; Gabriel, N.; Durbin-Johnson, B.; Hagerman, R.J.; Tassone, F. High MMP-9 Activity Levels in Fragile X Syndrome Are Lowered by Minocycline. Am. J. Med. Genet. Part A 2013, 161, 1897–1903. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R. Neuroimmune Interactions: From the Brain to the Immune System and Vice Versa. Physiol. Rev. 2018, 98, 477–504. [Google Scholar] [CrossRef] [PubMed]
- Prilutsky, D.; Kho, A.T.; Palmer, N.P.; Bhakar, A.L.; Smedemark-Margulies, N.; Kong, S.W.; Margulies, D.M.; Bear, M.F.; Kohane, I.S. Gene Expression Analysis in Fmr1KO Mice Identifies an Immunological Signature in Brain Tissue and MGluR5-Related Signaling in Primary Neuronal Cultures. Mol. Autism 2015, 6, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, S.L.; Nolan, S.O.; Taube, J.H.; Lugo, J.N. Adult Fmr1 Knockout Mice Present with Deficiencies in Hippocampal Interleukin-6 and Tumor Necrosis Factor-α Expression. Neuroreport 2017, 28, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Careaga, M.; Noyon, T.; Basuta, K.; Van de Water, J.; Tassone, F.; Hagerman, R.J.; Ashwood, P. Group I Metabotropic Glutamate Receptor Mediated Dynamic Immune Dysfunction in Children with Fragile X Syndrome. J. Neuroinflamm. 2014, 11, 110. [Google Scholar] [CrossRef] [Green Version]
- Van Dijck, A.; Barbosa, S.; Bermudez-Martin, P.; Khalfallah, O.; Gilet, C.; Martinuzzi, E.; Elinck, E.; Kooy, R.F.; Glaichenhaus, N.; Davidovic, L. Reduced Serum Levels of Pro-Inflammatory Chemokines in Fragile X Syndrome. BMC Neurol. 2020, 20, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashwood, P.; Nguyen, D.V.; Hessl, D.; Hagerman, R.J.; Tassone, F. Plasma Cytokine Profiles in Fragile X Subjects: Is There a Role for Cytokines in the Pathogenesis? Brain Behav. Immun. 2010, 24, 898–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.-H.; Palmer, N.; Fox, K.; Prock, L.; Mandl, K.D.; Kohane, I.S.; Prilutsky, D. The Phenotypical Implications of Immune Dysregulation in Fragile X Syndrome. Eur. J. Neurol. 2020, 27, 590–593. [Google Scholar] [CrossRef]
- Leboucher, A.; Pisani, D.F.; Martinez-Gili, L.; Chilloux, J.; Bermudez-Martin, P.; Van Dijck, A.; Ganief, T.; Macek, B.; Becker, J.A.J.; Le Merrer, J.; et al. The Translational Regulator FMRP Controls Lipid and Glucose Metabolism in Mice and Humans. Mol. Metab. 2019, 21, 22–35. [Google Scholar] [CrossRef]
- Davidovic, L.; Navratil, V.; Bonaccorso, C.M.; Catania, M.V.; Bardoni, B.; Dumas, M.-E. A Metabolomic and Systems Biology Perspective on the Brain of the Fragile X Syndrome Mouse Model. Genome Res. 2011, 21, 2190–2202. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and Regulation of Cholesterol Homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Turley, S.D. Thematic Review Series: Brain Lipids. Cholesterol Metabolism in the Central Nervous System during Early Development and in the Mature Animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef] [Green Version]
- Martín, M.G.; Pfrieger, F.; Dotti, C.G. Cholesterol in Brain Disease: Sometimes Determinant and Frequently Implicated. EMBO Rep. 2014, 15, 1036–1052. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Stransky, A.; Tierney, E. Cognitive and Behavioral Aspects of Smith–Lemli–Opitz Syndrome. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2012, 160C, 295–300. [Google Scholar] [CrossRef]
- Boland, M.R.; Tatonetti, N.P. Investigation of 7-Dehydrocholesterol Reductase Pathway to Elucidate off-Target Prenatal Effects of Pharmaceuticals: A Systematic Review. Pharm. J. 2016, 16, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Tierney, E.; Bukelis, I.; Thompson, R.E.; Ahmed, K.; Aneja, A.; Kratz, L.; Kelley, R.I. Abnormalities of Cholesterol Metabolism in Autism Spectrum Disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141B, 666–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.-K.; Neggers, Y.H.; Shin, C.-S.; Kim, E.; Kim, E.M. Alterations in Lipid Profile of Autistic Boys: A Case Control Study. Nutr. Res. 2010, 30, 255–260. [Google Scholar] [CrossRef]
- Benachenhou, S.; Etcheverry, A.; Galarneau, L.; Dubé, J.; Çaku, A. Implication of Hypocholesterolemia in Autism Spectrum Disorder and Its Associated Comorbidities: A Retrospective Case–Control Study. Autism Res. 2019, 12, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Levin, R.; Shah, H.; Mathur, S.; Darnell, J.C.; Ouyang, B. Cholesterol Levels in Fragile X Syndrome. Am. J. Med. Genet. 2015, 167, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Çaku, A.; Seidah, N.G.; Lortie, A.; Gagné, N.; Perron, P.; Dubé, J.; Corbin, F. New Insights of Altered Lipid Profile in Fragile X Syndrome. PLoS ONE 2017, 12, e0174301. [Google Scholar] [CrossRef] [Green Version]
- Ben Djoudi Ouadda, A.; Gauthier, M.-S.; Susan-Resiga, D.; Girard, E.; Essalmani, R.; Black, M.; Marcinkiewicz, J.; Forget, D.; Hamelin, J.; Evagelidis, A.; et al. Ser-Phosphorylation of PCSK9 (Proprotein Convertase Subtilisin-Kexin 9) by Fam20C (Family with Sequence Similarity 20, Member C) Kinase Enhances Its Ability to Degrade the LDLR (Low-Density Lipoprotein Receptor). ATVB 2019, 39, 1996–2013. [Google Scholar] [CrossRef] [PubMed]
- London, E.; Bloyd, M.; Stratakis, C.A. PKA Functions in Metabolism and Resistance to Obesity: Lessons from Mouse and Human Studies. J. Endocrinol. 2020, 246, R51–R64. [Google Scholar] [CrossRef] [PubMed]
- Conti, M. Phosphodiesterases and Cyclic Nucleotide Signaling in Endocrine Cells. Mol. Endocrinol. 2000, 14, 1317–1327. [Google Scholar] [CrossRef]
- Goldsmith, Z.G.; Dhanasekaran, D.N. G Protein Regulation of MAPK Networks. Oncogene 2007, 26, 3122–3142. [Google Scholar] [CrossRef] [Green Version]
- Dityatev, A.; El-Husseini, A. (Eds.) Molecular Mechanisms of Synaptogenesis; Springer: New York, NY, USA, 2006; ISBN 978-0-387-32560-6. [Google Scholar]
- Crawford, D.C.; Mennerick, S. Presynaptically Silent Synapses: Dormancy and Awakening of Presynaptic Vesicle Release. Neuroscientist 2012, 18, 216–223. [Google Scholar] [CrossRef]
- Beavo, J.A.; Brunton, L.L. Cyclic Nucleotide Research -- Still Expanding after Half a Century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Sklena, P. Demonstration of Abnormal Cyclic AMP Production in Platelets from Patients with Fragile X Syndrome. Am. J. Med. Genet. 1993, 45, 81–87. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Huttenlocher, P.R. Cyclic AMP Metabolism in Fragile X Syndrome. Ann. Neurol. 1992, 31, 22–26. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Hicar, M.; Ciurlionis, R. Reduced cyclic AMP production in fragile X syndrome: Cytogenetic and molecular correlations. Pediatr. Res. 1995, 38, 638–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry-Kravis, E.; Ciurlionis, R. Overexpression of Fragile X Gene (FMR-1) Transcripts Increases CAMP Production in Neural Cells. J. Neurosci. Res. 1998, 51, 41–48. [Google Scholar] [CrossRef]
- Kelley, D.J.; Davidson, R.J.; Elliott, J.L.; Lahvis, G.P.; Yin, J.C.P.; Bhattacharyya, A. The Cyclic AMP Cascade Is Altered in the Fragile X Nervous System. PLoS ONE 2007, 2, e931. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.; Bhattacharyya, A.; Lahvis, G.; Yin, J.; Malter, J.; Davidson, R. The Cyclic AMP Phenotype of Fragile X and Autism. Neurosci. Biobehav. Rev. 2008, 32, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.; Sardone, L.M.; Bonaccorso, C.M.; D’Antoni, S.; Spatuzza, M.; Gulisano, W.; Tropea, M.R.; Puzzo, D.; Leopoldo, M.; Lacivita, E.; et al. Activation of Serotonin 5-HT7 Receptors Modulates Hippocampal Synaptic Plasticity by Stimulation of Adenylate Cyclases and Rescues Learning and Behavior in a Mouse Model of Fragile X Syndrome. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Maurin, T.; Melancia, F.; Jarjat, M.; Castro, L.; Costa, L.; Delhaye, S.; Khayachi, A.; Castagnola, S.; Mota, E.; Di Giorgio, A.; et al. Involvement of Phosphodiesterase 2A Activity in the Pathophysiology of Fragile X Syndrome. Cereb. Cortex 2019, 29, 3241–3252. [Google Scholar] [CrossRef]
- Gurney, M.E.; Cogram, P.; Deacon, R.M.; Rex, C.; Tranfaglia, M. Multiple Behavior Phenotypes of the Fragile-X Syndrome Mouse Model Respond to Chronic Inhibition of Phosphodiesterase-4D (PDE4D). Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.H.; Schoenfeld, B.P.; Bell, A.J.; Hinchey, J.; Rosenfelt, C.; Gertner, M.J.; Campbell, S.R.; Emerson, D.; Hinchey, P.; Kollaros, M.; et al. Multiple Drug Treatments That Increase CAMP Signaling Restore Long-Term Memory and Aberrant Signaling in Fragile X Syndrome Models. Front. Behav. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumkötter, F.; Schmidt, N.; Vargas, C.; Schilling, S.; Weber, R.; Wagner, K.; Fiedler, S.; Klug, W.; Radzimanowski, J.; Nickolaus, S.; et al. Amyloid Precursor Protein Dimerization and Synaptogenic Function Depend on Copper Binding to the Growth Factor-like Domain. J. Neurosci. 2014, 34, 11159–11172. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.V. β-Amyloid Precursor Protein (APP) and the Human Diseases. Aims Neurosci. 2019, 6, 273–281. [Google Scholar] [CrossRef]
- Lee, E.K.; Kim, H.H.; Kuwano, Y.; Abdelmohsen, K.; Srikantan, S.; Subaran, S.S.; Gleichmann, M.; Mughal, M.R.; Martindale, J.L.; Yang, X.; et al. HnRNP C Promotes APP Translation by Competing with FMRP for APP MRNA Recruitment to P Bodies. Nat. Struct. Mol. Biol. 2010, 17, 732–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasciuto, E.; Ahmed, T.; Wahle, T.; Gardoni, F.; D’Andrea, L.; Pacini, L.; Jacquemont, S.; Tassone, F.; Balschun, D.; Dotti, C.G.; et al. Dysregulated ADAM10-Mediated Processing of APP during a Critical Time Window Leads to Synaptic Deficits in Fragile X Syndrome. Neuron 2015, 87, 382–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westmark, C.J.; Malter, J.S. FMRP Mediates MGluR5-Dependent Translation of Amyloid Precursor Protein. PLoS Biol. 2007, 5. [Google Scholar] [CrossRef] [PubMed]
- Westmark, C.J.; Westmark, P.R.; O’Riordan, K.J.; Ray, B.C.; Hervey, C.M.; Salamat, M.S.; Abozeid, S.H.; Stein, K.M.; Stodola, L.A.; Tranfaglia, M.; et al. Reversal of Fragile X Phenotypes by Manipulation of AβPP/Aβ Levels in Fmr1KO Mice. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, D.K.; Chen, D.; Farlow, M.R.; Dunn, D.W.; Maloney, B.; Zimmer, J.A.; Lahiri, D.K. High Levels of Alzheimer Beta-Amyloid Precursor Protein (APP) in Children With Severely Autistic Behavior and Aggression. J. Child Neurol. 2006, 21, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.; Long, J.M.; Sokol, D.K.; Lahiri, D.K. Increased Secreted Amyloid Precursor Protein-α (SAPPα) in Severe Autism: Proposal of a Specific, Anabolic Pathway and Putative Biomarker. PLoS ONE 2011, 6, e20405. [Google Scholar] [CrossRef] [Green Version]
- Ray, B.; Sokol, D.K.; Maloney, B.; Lahiri, D.K. Finding Novel Distinctions between the SAPPα-Mediated Anabolic Biochemical Pathways in Autism Spectrum Disorder and Fragile X Syndrome Plasma and Brain Tissue. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- McLane, R.D.; Schmitt, L.M.; Pedapati, E.V.; Shaffer, R.C.; Dominick, K.C.; Horn, P.S.; Gross, C.; Erickson, C.A. Peripheral Amyloid Precursor Protein Derivative Expression in Fragile X Syndrome. Front. Integr. Neurosci. 2019, 13, 49. [Google Scholar] [CrossRef]
- Erickson, C.A.; Ray, B.; Maloney, B.; Wink, L.K.; Bowers, K.; Schaefer, T.L.; McDougle, C.J.; Sokol, D.K.; Lahiri, D.K. Impact of Acamprosate on Plasma Amyloid-β Precursor Protein in Youth: A Pilot Analysis in Fragile X Syndrome-Associated and Idiopathic Autism Spectrum Disorder Suggests a Pharmacodynamic Protein Marker. J. Psychiatr. Res. 2014, 59, 220–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic representation of the different “Omic” categories: (A) a simplistic “Omic” representation in which a gene encodes a single mRNA, which is further translated in a unique protein that can ultimately exert its biological activity, which in this example produces a metabolite. (B) A large diversity of biomolecules exists within each “Omic” category. (C) Schematization of the different interaction between component of each “Omic” categories.
Figure 1.
Schematic representation of the different “Omic” categories: (A) a simplistic “Omic” representation in which a gene encodes a single mRNA, which is further translated in a unique protein that can ultimately exert its biological activity, which in this example produces a metabolite. (B) A large diversity of biomolecules exists within each “Omic” category. (C) Schematization of the different interaction between component of each “Omic” categories.

Figure 2.
Genetics of fragile X syndrome: (A) CGG trinucleotide repeats are found between the promoter and the first exon of the FMR1 gene. A normal allele has fewer than 55 while a fully mutated one has more than 200. A full mutation leads to the silencing of the FMR1 gene throughout an epigenetic mechanism which is in part characterized by the methylation (Me) of the gene. A pre-mutation allele has between 55 and 200 CGG repeats and can lead to FXTAS (Fragile X Tremor and Ataxia Syndrome) or FXPOI (Fragile X Primary Ovarian Insufficiency). (B) Two types of mosaicism are observed in Fragile X. Expansion mosaicism is characterized by a cell population carrying a pre-mutation and another with a fully mutated and methylated allele. On the other hand, methylation mosaicism is characterized by two populations of fully mutated cells but only one of them carries a methylated allele. Both types of mosaicism result in low FMRP expression.
Figure 2.
Genetics of fragile X syndrome: (A) CGG trinucleotide repeats are found between the promoter and the first exon of the FMR1 gene. A normal allele has fewer than 55 while a fully mutated one has more than 200. A full mutation leads to the silencing of the FMR1 gene throughout an epigenetic mechanism which is in part characterized by the methylation (Me) of the gene. A pre-mutation allele has between 55 and 200 CGG repeats and can lead to FXTAS (Fragile X Tremor and Ataxia Syndrome) or FXPOI (Fragile X Primary Ovarian Insufficiency). (B) Two types of mosaicism are observed in Fragile X. Expansion mosaicism is characterized by a cell population carrying a pre-mutation and another with a fully mutated and methylated allele. On the other hand, methylation mosaicism is characterized by two populations of fully mutated cells but only one of them carries a methylated allele. Both types of mosaicism result in low FMRP expression.

Figure 3.
Schematization of FXS physiopathology, resulting from an imbalance between excitatory (glutamate) and inhibitory (GABA) neurotransmission. (A) Activation of GABA receptors at the presynaptic neurons inhibits the release of glutamate into the synaptic cleft. Glutamate stimulation of mGluR5 at the postsynaptic neuron leads to the activation of many downstream effectors, mainly the Ras/Raf/Mek/ERK and PI3K/AKT/mTOR signaling cascades. These pathways lead, among other things, to the translation of proteins which promote LTD and impair LTP. (B) GABA output is decreased in the absence of FMRP, resulting in an improper neuronal inhibition. The resulting enhanced glutamate release promotes the activation of mGluR5, which ultimately lead to an exaggerated LTD and a reduced LTP.
Figure 3.
Schematization of FXS physiopathology, resulting from an imbalance between excitatory (glutamate) and inhibitory (GABA) neurotransmission. (A) Activation of GABA receptors at the presynaptic neurons inhibits the release of glutamate into the synaptic cleft. Glutamate stimulation of mGluR5 at the postsynaptic neuron leads to the activation of many downstream effectors, mainly the Ras/Raf/Mek/ERK and PI3K/AKT/mTOR signaling cascades. These pathways lead, among other things, to the translation of proteins which promote LTD and impair LTP. (B) GABA output is decreased in the absence of FMRP, resulting in an improper neuronal inhibition. The resulting enhanced glutamate release promotes the activation of mGluR5, which ultimately lead to an exaggerated LTD and a reduced LTP.

Figure 4.
Summary of some of the various biochemicals alterations composing the fragile X phenotype on each “Omic” category.
Figure 4.
Summary of some of the various biochemicals alterations composing the fragile X phenotype on each “Omic” category.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dionne, O.; Corbin, F. An “Omic” Overview of Fragile X Syndrome. Biology 2021, 10, 433. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10050433
AMA Style
Dionne O, Corbin F. An “Omic” Overview of Fragile X Syndrome. Biology. 2021; 10(5):433. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10050433
Chicago/Turabian StyleDionne, Olivier, and François Corbin. 2021. "An “Omic” Overview of Fragile X Syndrome" Biology 10, no. 5: 433. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10050433
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.