
Improving Genomic Prediction with Machine Learning Incorporating TPE for Hyperparameters Optimization
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.3. Assessing Prediction Performance
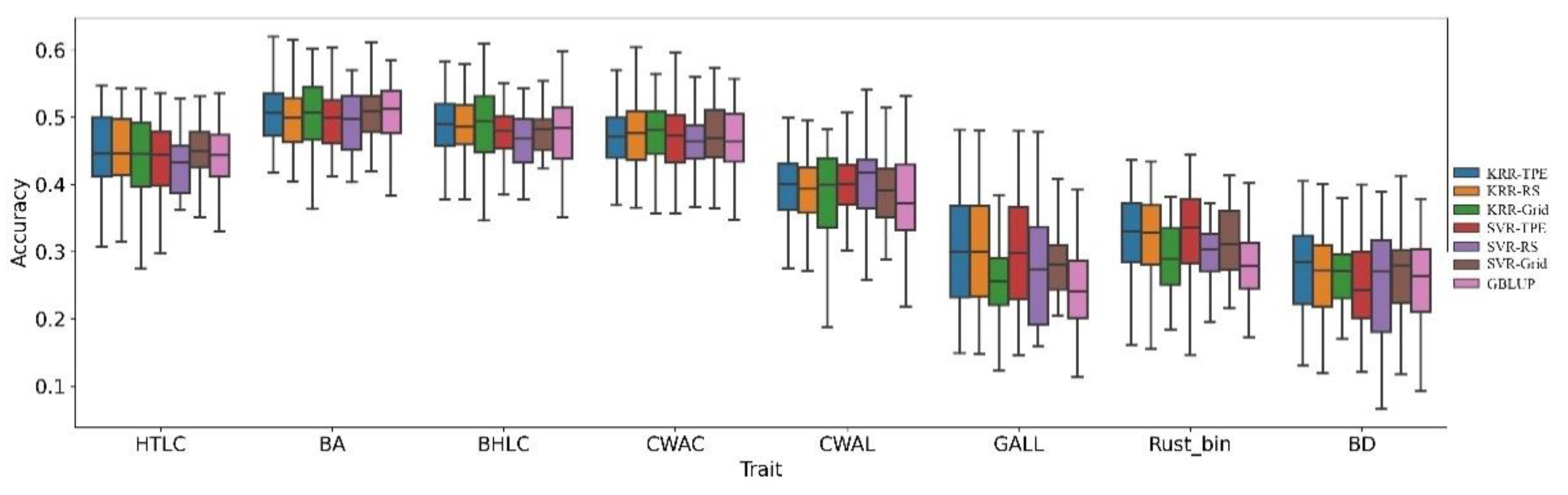
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Code Availability
References
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.J.; Goddard, M.E. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, B.J.; Visscher, P.M.; Goddard, M.E. Increased accuracy of artificial selection by using the realized relationship matrix. Genet. Res. 2009, 91, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, R.; Mucha, S.; Larroque, H.; Mcewan, J.; Conington, J. Genomic application in sheep and goat breeding. Anim. Front. 2016, 6, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Tusell, L.; Pérez-Rodríguez, P.; Forni, S.; Wu, X.L.; Gianola, D. Genome-enabled methods for predicting litter size in pigs: A comparison. Animal 2013, 7, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Stock, K.F.; Jönsson, L.; Ricard, A.; Mark, T. Genomic applications in horse breeding. Anim. Front. 2016, 6, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Kranis, A.; Gheyas, A.A.; Boschiero, C.; Turner, F.; Le, Y.; Smith, S.; Talbot, R.; Pirani, A.; Brew, F.; Kaiser, P. Development of a high density 600K SNP genotyping array for chicken. BMC Genom. 2013, 14, 59. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Gowda, M.; Liu, W.; Würschum, T.; Reif, J.C. Accuracy of genomic selection in European maize elite breeding Populations. Theor. Appl. Genet. 2011, 124, 769–776. [Google Scholar] [CrossRef]
- Poland, J.; Endelman, J.; Da Wson, J.; Rutkoski, J.; Wu, S.; Manes, Y.; Dreisigacker, S.; Crossa, J.; Sanchez-Villeda, H.; Sorrells, M. Genomic Selection in Wheat Breeding using Genotyping-by-Sequencing. Plant Genome 2012, 5, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Grenier, C.; Cao, T.V.; Ospina, Y.; Quintero, C.; Châtel, M.H.; Tohme, J.; Ahmadi, N. Accuracy of Genomic Selection in a Rice Synthetic Population Developed for Recurrent Selection Breeding. PLoS ONE 2015, 10, e0154976. [Google Scholar] [CrossRef]
- Vanraden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwissen, T.H.; Solberg, T.R.; Shepherd, R.; Woolliams, J.A. A fast algorithm for BayesB type of prediction of genome-wide estimates of genetic value. Genet. Sel. Evol. 2009, 41, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianola, D.; Okut, H.; Weigel, K.A.; Rosa, G.J. Predicting complex quantitative traits with Bayesian neural networks: A case study with Jersey cows and wheat. BMC Genet. 2011, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, A.; Da, C.; Tiago, B.; Alves, F.; Rafael, E.; Frossard, R.; Roberto, C.; Galvo, D. Genome-wide prediction for complex traits under the presence of dominance effects in simulated populations using GBLUP and machine learning methods. J. Anim. Sci. 2020, 98, skaa179. [Google Scholar]
- Ornella, L.; Gonzalez-Camacho, J.M.; Dreisigacker, S.; Crossa, J. Applications of genomic selection in breeding wheat for rust resistance. In Wheat Rust Diseases; Springer: Berlin/Heidelberg, Germany, 2017; pp. 173–182. [Google Scholar]
- Ghafouri, F.; Alipour, S.; Mohamadian Jeshvaghani, S. Application of machine learning approach and its subset algorithms in estimating genomic breeding values. Prof. J. Domest. 2020, 20, 19–29. [Google Scholar]
- Mitchell, T.M. Machine Learning; McGraw-Hill: New York, NY, USA, 2003. [Google Scholar]
- Ansari, M.F.; Alankarkaur, B.; Kaur, H. A Prediction of Heart Disease Using Machine Learning Algorithms. In Proceedings of the ICIPCN: International Conference on Image Processing and Capsule Networks, Bangkok, Thailand, 27–28 May 2021. [Google Scholar]
- Austin, P.C.; Tu, J.V.; Ho, J.E.; Levy, D.; Lee, D.S. Using methods from the data-mining and machine-learning literature for disease classification and prediction: A case study examining classification of heart failure subtypes. J. Clin. Epidemiol. 2013, 66, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Shammut, M. Driverless Cars: A Historical Overview. 2020.
- Mohammed, A.A.; Minhas, R.; Wu, Q.; Sid-Ahmed, M.A. Human face recognition based on multidimensional PCA and extreme learning machine. Pattern Recognit. 2012, 44, 2588–2597. [Google Scholar] [CrossRef]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Hassabis, D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Kolosov, N.; Daly, M.J.; Artomov, M. Prioritization of disease genes from GWAS using ensemble-based positive-unlabeled learning. Eur. J. Hum. 2021, 29, 1527–1535. [Google Scholar] [CrossRef]
- Hao, T.; Nikoloski, Z. Machine learning approaches for crop improvement: Leveraging phenotypic and genotypic big data. J. Plant Physiol. 2021, 257, 153354. [Google Scholar]
- Hao, T.; Küken, A.; Nikoloski, Z. Integrating molecular markers into metabolic models improves genomic selection for Arabidopsis growth. Nat. Commun. 2020, 11, 2410. [Google Scholar]
- Manuel, G.; Leonardo, O.; Paulino, P.R.; Daniel, G.; Susanne, D.; José, C. Applications of Machine Learning Methods to Genomic Selection in Breeding Wheat for Rust Resistance. Plant Genome 2018, 11, 170104. [Google Scholar]
- Okut, H.; Wu, X.L.; Rosa, G.J.; Bauck, S.; Woodward, B.W.; Schnabel, R.D.; Taylor, J.F.; Gianola, D. Predicting expected progeny difference for marbling score in Angus cattle using artificial neural networks and Bayesian regression models. Genet. Sel. Evol. 2013, 45, 34. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-López, O.A.; Martín-Vallejo, J.; Crossa, J.; Gianola, D.; Hernández-Suárez, C.M.; Montesinos-López, A.; Juliana, P.; Singh, R. A benchmarking between deep learning, support vector machine and Bayesian threshold best linear unbiased prediction for predicting ordinal traits in plant breeding. G3 Genes Genomes Genet. 2019, 9, 601–618. [Google Scholar] [CrossRef]
- Ozaki, Y.; Tanigaki, Y.; Watanabe, S.; Onishi, M. Multiobjective tree-structured parzen estimator for computationally expensive optimization problems. In Proceedings of the 2020 Genetic and Evolutionary Computation Conference, Cancún, Mexico, 8–12 July 2020; pp. 533–541. [Google Scholar]
- Nguyen, H.-P.; Liu, J.; Zio, E. A long-term prediction approach based on long short-term memory neural networks with automatic parameter optimization by Tree-structured Parzen Estimator and applied to time-series data of NPP steam generators. Appl. Soft Comput. 2020, 89, 106116. [Google Scholar] [CrossRef] [Green Version]
- Erwianda, M.S.F.; Kusumawardani, S.S.; Santosa, P.I.; Rimadana, M.R. Improving confusion-state classifier model using xgboost and tree-structured parzen estimator. In Proceedings of the 2019 International Seminar on Research of Information Technology and Intelligent Systems (ISRITI), Yogyakarta, Indonesia, 5–6 December 2019; pp. 309–313. [Google Scholar]
- Shen, K.; Qin, H.; Zhou, J.; Liu, G. Runoff Probability Prediction Model Based on Natural Gradient Boosting with Tree-Structured Parzen Estimator Optimization. Water 2022, 14, 545. [Google Scholar] [CrossRef]
- Rasmussen, C.E.; Williams, C. Gaussian processes for machine learning the mit press. Camb. MA 2006, 32, 68. [Google Scholar]
- Usai, M.G.; Gaspa, G.; Macciotta, N.P.; Carta, A.; Casu, S. XVI th QTLMAS: Simulated dataset and comparative analysis of submitted results for QTL mapping and genomic evaluation. BMC Proc. 2014, 8, S1. [Google Scholar] [CrossRef] [Green Version]
- Zhe, Z.; Erbe, M.; He, J.; Ober, U.; Li, J. Accuracy of Whole-Genome Prediction Using a Genetic Architecture-Enhanced Variance-Covariance Matrix. G3 Genes Genomes Genet. 2015, 5, 615–627. [Google Scholar]
- Li, H.; Su, G.; Jiang, L.; Bao, Z. An efficient unified model for genome-wide association studies and genomic selection. Genet. Sel. Evol. 2017, 49, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.; Bender, D.; Maller, J.; Sklar, P.; Bakker, P.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Eckert, A.J.; Van Heerwaarden, J.; Wegrzyn, J.L.; Nelson, C.D.; Ross-Ibarra, J.; Gonzalez-Martinez, S.C.; Neale, D.B. Patterns of population structure and environmental associations to aridity across the range of loblolly pine (Pinus taeda L., Pinaceae). Genetics 2010, 185, 969. [Google Scholar] [CrossRef] [Green Version]
- Cleveland, M.A.; Hickey, J.M.; Forni, S. A common dataset for genomic analysis of livestock populations. G3 Genes Genomes Genet. 2012, 2, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.L.; Park, C.A.; Wu, X.L.; Reecy, J.M. Animal QTLdb: An improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013, 41, D871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhe, Z.; Ober, U.; Erbe, M.; Hao, Z.; Gao, N. Improving the Accuracy of Whole Genome Prediction for Complex Traits Using the Results of Genome Wide Association Studies. PloS ONE 2014, 9, e93017. [Google Scholar]
- Henderson, C.R. Best linear unbiased estimation and prediction under a selection model. Biometrics 1975, 31, 423–447. [Google Scholar] [CrossRef] [Green Version]
- Habier, D.; Fernando, R.L.; Dekkers, J.C.M. The Impact of Genetic Relationship Information on Genome-Assisted Breeding Values. Genetics 2007, 177, 2389–2397. [Google Scholar] [CrossRef]
- Rong, G.; Li, K.; Su, Y.; Tong, Z.; Liu, X.; Zhang, J.; Zhang, Y.; Li, T. Comparison of Tree-Structured Parzen Estimator Optimization in Three Typical Neural Network Models for Landslide Susceptibility Assessment. Remote Sens. 2021, 13, 4694. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Trait | N | h2 | Mean | SD |
|---|---|---|---|---|---|
| Simulation | T1 | 3000 | 0.36 | 0.00 | 176.52 |
| T2 | 3000 | 0.35 | 0.00 | 9.51 | |
| T3 | 3000 | 0.52 | 0.00 | 0.02 | |
| Beef cattle | LW | 1285 | 0.30 | 510.37 | 73.50 |
| ADG | 1282 | 0.28 | 0.97 | 0.21 | |
| NMW | 1273 | 0.32 | 233.02 | 41.75 | |
| TT | 1275 | 0.30 | 17.89 | 2.12 | |
| TW | 1284 | 0.29 | 8.75 | 1.98 | |
| EMW | 1281 | 0.38 | 10.67 | 2.22 | |
| Loblolly pine | HTLC | 861 | 0.31 | 20.30 | 73.31 |
| BA | 861 | 0.45 | 2.28 | 42.03 | |
| BD | 910 | 0.11 | 0.05 | 1.20 | |
| BHLC | 861 | 0.35 | 0.092 | 0.51 | |
| CWAC | 861 | 0.45 | 2.28 | 42.03 | |
| CWAL | 861 | 0.27 | 2.44 | 27.33 | |
| GALL | 807 | 0.12 | −0.022 | 1.31 | |
| Rust_bin | 807 | 0.21 | −0.014 | 0.40 | |
| Pig | T2 | 2715 | 0.16 | 0.00 | 1.12 |
| T3 | 3141 | 0.22 | 0.71 | 0.96 | |
| T4 | 3152 | 0.32 | −1.07 | 2.33 | |
| T5 | 3184 | 0.38 | 37.99 | 60.45 | |
| German Holstein | MY | 5024 | 0.95 | 370.79 | 641.60 |
| MFP | 5024 | 0.94 | −0.06 | 0.28 | |
| SCS | 5024 | 0.88 | 102.32 | 11.73 |
| KRR | SVR | ||
|---|---|---|---|
| Kernel | Cosine, RBF, Linear | Kernel | RBF, Linear, Ploy |
| Gamma | 0.000001–0.001 | Degree | 1, 2, 3, 4 |
| Alpha | 0–10 | Gamma | 0.000001–0.001 |
| K | 1–n | C | 0.1–100 |
| K | 1–n | ||
| Trait | KRR | SVR | GBLUP | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TPE | RS | Grid | TPE | RS | Grid | |||||
| T1 | 0.402 | 0.400 | 0.394 | 0.39 | 0.386 | 0.393 | 0.406 | |||
| T2 | 0.398 | 0.395 | 0.395 | 0.387 | 0.382 | 0.384 | 0.400 | |||
| T3 | 0.546 | 0.541 | 0.544 | 0.545 | ||||||
| Trait | KRR | SVR | GBLUP | ||||
|---|---|---|---|---|---|---|---|
| TPE | RS | Grid | TPE | RS | Grid | ||
| LW | 0.298 | 0.288 | 0.264 | 0.278 | 0.271 | 0.286 | 0.276 |
| ADG | 0.274 | 0.275 | 0.252 | 0.215 | 0.202 | 0.222 | 0.265 |
| MW | 0.298 | 0.287 | 0.27 | 0.268 | 0.271 | 0.262 | 0.270 |
| TT | 0.294 | 0.295 | 0.26 | 0.249 | 0.288 | 0.247 | 0.274 |
| TW | 0.208 | 0.196 | 0.197 | 0.193 | 0.201 | 0.185 | 0.193 |
| EMW | 0.314 | 0.316 | 0.288 | 0.291 | 0.286 | 0.27 | 0.295 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, M.; An, B.; Li, K.; Du, L.; Deng, T.; Cao, S.; Du, Y.; Xu, L.; Gao, X.; Zhang, L.; et al. Improving Genomic Prediction with Machine Learning Incorporating TPE for Hyperparameters Optimization. Biology 2022, 11, 1647. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11111647
Liang M, An B, Li K, Du L, Deng T, Cao S, Du Y, Xu L, Gao X, Zhang L, et al. Improving Genomic Prediction with Machine Learning Incorporating TPE for Hyperparameters Optimization. Biology. 2022; 11(11):1647. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11111647
Chicago/Turabian StyleLiang, Mang, Bingxing An, Keanning Li, Lili Du, Tianyu Deng, Sheng Cao, Yueying Du, Lingyang Xu, Xue Gao, Lupei Zhang, and et al. 2022. "Improving Genomic Prediction with Machine Learning Incorporating TPE for Hyperparameters Optimization" Biology 11, no. 11: 1647. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11111647