
Single Gene Mutations in Pkd1 or Tsc2 Alter Extracellular Vesicle Production and Trafficking

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Pkd1 Mutant Renal Epithelial Cells Produce More EVs Than Pkd2 Mutant Cells
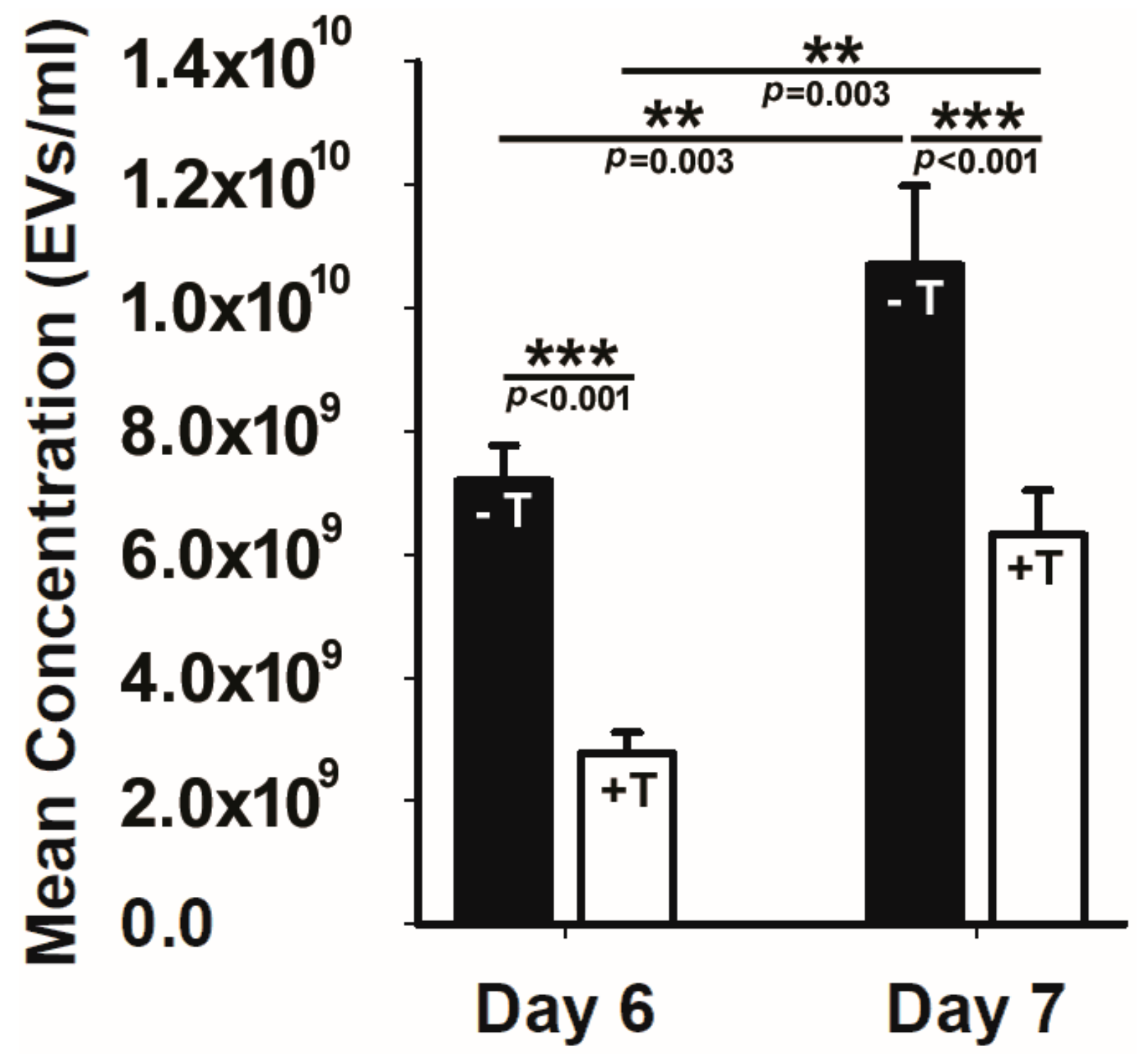
2.2. Cilial Disruption Reduces EV Production
2.3. Renal EVs Home to the Kidney
2.4. EVs from Cultured Clonal Renal Epithelial Cell Home to Kidney
2.5. Single Gene Defects Alter Cellular EV Uptake
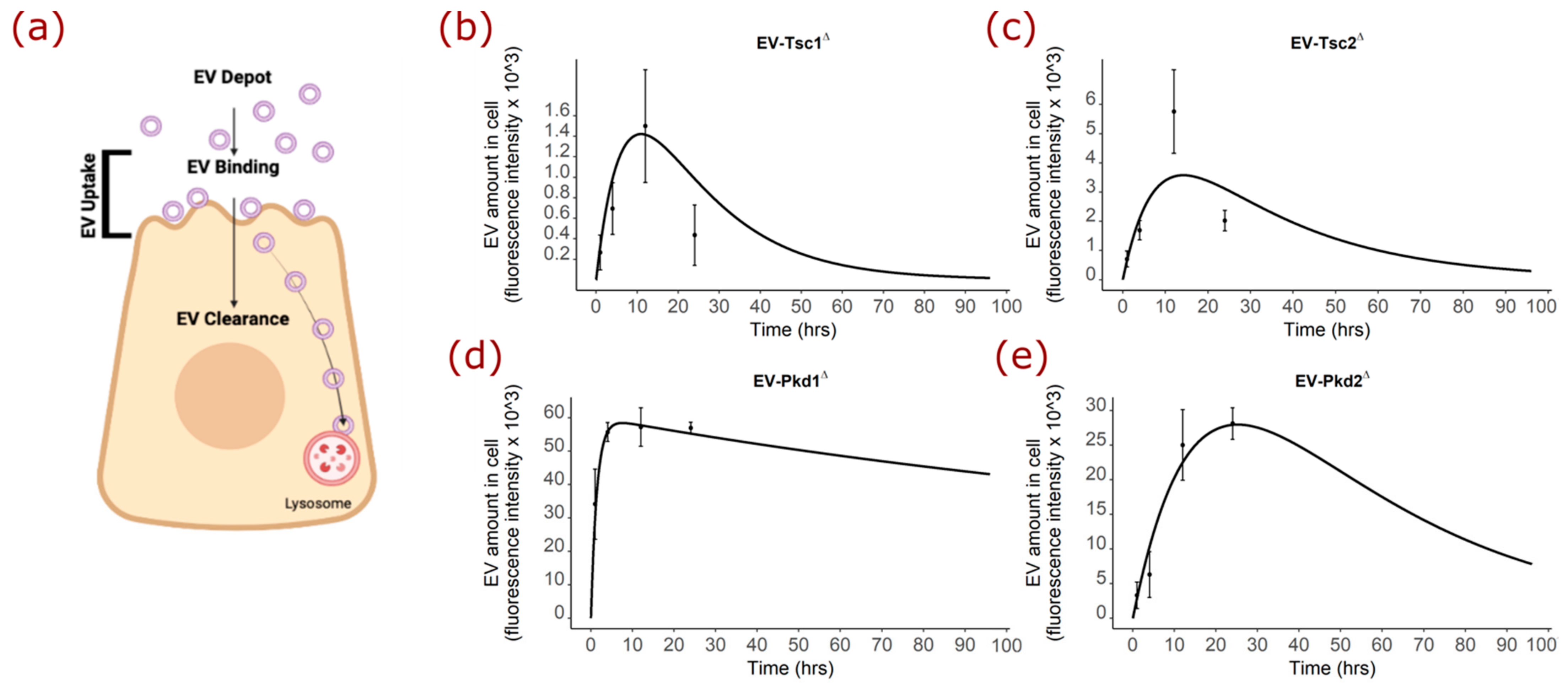
2.6. Mathematical Modeling of the EV Staining Results Offer Insight into Polycystin-1 Function
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animals
4.3. EV Isolation
4.3.1. From Cell Culture Media
4.3.2. From Mouse Cyst Fluid
4.3.3. From Human Cyst Fluid
4.3.4. From Mouse Kidney Tissue
4.4. EV Isolation and Column Setup and Operation
4.5. Physical Characterization of Extracellular Vesicles
4.6. EV Marker Analysis by Western Blot
4.7. EV Labelling
4.8. EV Trafficking In Vitro
4.9. EV Trafficking Ex Vivo
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- USRDS. USRDS Annual Data Report. Available online: https://adr.usrds.org/2020/reference-tables (accessed on 4 August 2021).
- Chapman, A.B.; Devuyst, O.; Eckardt, K.-U.U.; Gansevoort, R.T.; Harris, T.; Horie, S.; Kasiske, B.L.; Odland, D.; Pei, Y.; Perrone, R.D.; et al. Autosomal-dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordido, A.; Vizoso-Gonzalez, M.; Garcia-Gonzalez, M. Molecular Pathophysiology of Autosomal Recessive Polycystic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 6523. [Google Scholar] [CrossRef] [PubMed]
- Bissler, J.J.; Kingswood, J.C. Renal manifestation of tuberous sclerosis complex. Am. J. Med Genet. Part C: Semin. Med Genet. 2018, 178, 338–347. [Google Scholar] [CrossRef]
- Ciullo, A.; Li, C.; Li, L.; Ungerleider, K.C.; Peck, K.; Marbán, E.; Ibrahim, A.G. Biodistribution of unmodified cardiosphere-derived cell extracellular vesicles using single RNA tracing. J. Extracell. Vesicles 2022, 11, e12178. [Google Scholar] [CrossRef] [PubMed]
- European Chromosome 16 Tuberous Sclerosis Consortium Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993, 75, 1305–1315. [CrossRef]
- Brook-Carter, P.T.; Peral, B.; Ward, C.J.; Thompson, P.; Hughes, J.; Maheshwar, M.M.; Nellist, M.; Gamble, V.; Harris, P.C.; Sampson, J.R. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease--a contiguous gene syndrome. Nat Genet 1994, 8, 328–332. [Google Scholar] [CrossRef]
- Cabrera-López, C.; Bullich, G.; Martí, T.; Català, V.; Ballarín, J.; Bissler, J.J.J.; Harris, P.C.P.C.; Ars, E.; Torra, R. Insight into response to mTOR inhibition when PKD1 and TSC2 are mutated. BMC Med. Genet. 2015, 16, 39. [Google Scholar] [CrossRef] [Green Version]
- Lantinga-van Leeuwen, I.S.; Dauwerse, J.G.; Baelde, H.J.; Leonhard, W.N.; van de Wal, A.; Ward, C.J.; Verbeek, S.; Deruiter, M.C.; Breuning, M.H.; de Heer, E.; et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum. Mol. Genet. 2004, 13, 3069–3077. [Google Scholar] [CrossRef] [Green Version]
- Reeders, S.T. Multilocus polycystic disease. Nat. Genet. 1992, 1, 235–237. [Google Scholar] [CrossRef]
- Qian, F.; Watnick, T.J.; Onuchic, L.F.; Germino, G.G. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell 1996, 87, 979–987. [Google Scholar] [CrossRef] [Green Version]
- Brasier, J.L.; Henske, E.P. Loss of the polycystic kidney disease (PKD1) region of chromosome 16p13 in renal cyst cells supports a loss-of-function model for cyst pathogenesis. J. Clin. Investig. 1997, 99, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koptides, M.; Constantinides, R.; Kyriakides, G.; Hadjigavriel, M.; Patsalis, P.C.; Pierides, A.; Deltas, C.C. Loss of heterozygosity in polycystic kidney disease with a missense mutation in the repeated region of PKD1. Hum. Genet. 1998, 103, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Watnick, T.; He, N.; Wang, K.; Liang, Y.; Parfrey, P.; Germino, G.; St George-Hyslop, P. Somatic PKD2 mutations in individual kidney and liver cysts support a "two-hit" model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1999, 10, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Badenas, C.; Torra, R.; Pérez-Oller, L.; Mallolas, J.; Talbot-Wright, R.; Torregrosa, V.; Darnell, A. Loss of heterozygosity in renal and hepatic epithelial cystic cells from ADPKD1 patients. Eur. J. Hum. Genet. 2000, 8, 487–492. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 50. [Google Scholar] [CrossRef]
- Kim, I.; Li, C.; Liang, D.; Chen, X.Z.; Coffy, R.J.; Ma, J.; Zhao, P.; Wu, G. Polycystin-2 expression is regulated by a PC2-binding domain in the intracellular portion of fibrocystin. J. Biol. Chem. 2008, 283, 31559–31566. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.T.; Chiou, Y.Y.; Wang, E.; Lin, H.K.; Lin, Y.T.; Chi, Y.C.; Wang, C.K.L.; Tang, M.J.; Li, H. Defining a link with autosomal-dominant polycystic kidney disease in mice with congenitally low expression of Pkd1. Am. J. Pathol. 2006, 168, 205–220. [Google Scholar] [CrossRef] [Green Version]
- Lantinga-van Leeuwen, I.S.; Leonhard, W.N.; van der Wal, A.; Breuning, M.H.; de Heer, E.; Peters, D.J.M. Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum. Mol. Genet. 2007, 16, 3188–3196. [Google Scholar] [CrossRef] [Green Version]
- Leonhard, W.N.; Zandbergen, M.; Veraar, K.; Van Den Berg, S.; Van Der Weerd, L.; Breuning, M.; De Heer, E.; Peters, D.J.M. Scattered deletion of PKD1 in kidneys causes a cystic snowball effect and recapitulates polycystic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1322–1333. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, E.C.; Audrézet, M.P.; Le Meur, Y.; Chen, J.M.; Férec, C. Genetics and pathogenesis of autosomal dominant polycystic kidney disease: 20 years on. Hum. Mutat. 2014, 35, 1393–1406. [Google Scholar] [CrossRef]
- Lam, H.C.; Nijmeh, J.; Henske, E.P. New developments in the genetics and pathogenesis of tumours in tuberous sclerosis complex. J. Pathol. 2017, 241, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannikou, K.; Malinowska, I.A.; Pugh, T.J.; Yan, R.; Tseng, Y.Y.; Oh, C.; Kim, J.; Tyburczy, M.E.; Chekaluk, Y.; Liu, Y.; et al. Whole Exome Sequencing Identifies TSC1/TSC2 Biallelic Loss as the Primary and Sufficient Driver Event for Renal Angiomyolipoma Development. PLoS Genet. 2016, 12, e1006242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onda, H.; Lueck, A.; Marks, P.W.; Warren, H.B.; Kwiatkowski, D.J. Tsc2(+/−) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J. Clin. Investig. 1999, 104, 687–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.; Bonnet, C.; Guy, C.; Idziaszczyk, S.; Colley, J.; Humphreys, V.; Maynard, J.; Sampson, J.R.; Cheadle, J.P. Tsc1 haploinsufficiency without mammalian target of rapamycin activation is sufficient for renal cyst formation in Tsc1+/− mice. Cancer Res. 2006, 66, 7934–7938. [Google Scholar] [CrossRef] [Green Version]
- Bonsib, S.M.; Boils, C.; Gokden, N.; Grignon, D.; Gu, X.; Higgins, J.P.T.; Leroy, X.; McKenney, J.K.; Nasr, S.H.; Phillips, C.; et al. Tuberous sclerosis complex: Hamartin and tuberin expression in renal cysts and its discordant expression in renal neoplasms. Pathol. Res. Pract. 2016, 212, 972–979. [Google Scholar] [CrossRef] [Green Version]
- Bacallao, R.L.; Mcneill, H. Cystic kidney diseases and planar cell polarity signaling. Clin. Genet. 2009, 75, 107–117. [Google Scholar] [CrossRef]
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893. [Google Scholar] [CrossRef]
- Barroso-Gil, M.; Olinger, E.; Sayer, J.A. Molecular genetics of renal ciliopathies. Biochem. Soc. Trans. 2021, 49, 1205–1220. [Google Scholar] [CrossRef]
- Kunimoto, K.; Bayly, R.D.; Vladar, E.K.; Vonderfecht, T.; Gallagher, A.-R.; Axelrod, J.D. Disruption of Core Planar Cell Polarity Signaling Regulates Renal Tubule Morphogenesis but Is Not Cystogenic. Curr. Biol. 2017, 27, 3120–3131.e4. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, C.S.; Aldred, M.; von Ruhland, C.; Harris, R.; Sandford, R.; Cheadle, J.P. Defects in cell polarity underlie TSC and ADPKD-associated cystogenesis. Hum. Mol. Genet. 2009, 18, 2166–2176. [Google Scholar] [CrossRef]
- Hartman, T.R.; Liu, D.; Zilfou, J.T.; Robb, V.; Morrison, T.; Watnick, T.; Henske, E.P. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum. Mol. Genet. 2009, 18, 151–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEneaney, L.J.; Tee, A.R. Finding a cure for tuberous sclerosis complex: From genetics through to targeted drug therapies. Adv. Genet. 2019, 103, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Belibi, F.; Ravichandran, K.; Zafar, I.; He, Z.; Edelstein, C.L. mTORC1/2 and rapamycin in female Han:SPRD rats with polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 2010, 300, F236–F244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA. 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.G.; Weimbs, T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J. Am. Soc. Nephrol. 2010, 21, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Pema, M.; Drusian, L.; Chiaravalli, M.; Castelli, M.; Yao, Q.; Ricciardi, S.; Somlo, S.; Qian, F.; Biffo, S.; Boletta, A. MTORC1-mediated inhibition of polycystin-1 expression drives renal cyst formation in tuberous sclerosis complex. Nat. Commun. 2016, 7, 10786. [Google Scholar] [CrossRef]
- Boletta, A. Emerging evidence of a link between the polycystins and the mTOR pathways. Pathogenetics 2009, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Wood, C.R.; Rosenbaum, J.L. Ciliary ectosomes: Transmissions from the cell’s antenna. Trends Cell Biol. 2015, 25, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Wood, C.R.; Huang, K.; Diener, D.R.; Rosenbaum, J.L. The Cilium Secretes Bioactive Ectosomes. Curr. Biol. 2013, 23, 906–911. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Silva, M.; Haas, L.A.; Morsci, N.S.; Nguyen, K.C.Q.Q.; Hall, D.H.; Barr, M.M.C. elegans ciliated sensory neurons release extracellular vesicles that function in animal communication. Curr. Biol. 2014, 24, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Pampliega, O.; Orhon, I.; Patel, B.; Sridhar, S.; Díaz-Carretero, A.; Beau, I.; Codogno, P.; Satir, B.H.; Satir, P.; Cuervo, A.M. Functional interaction between autophagy and ciliogenesis. Nature 2013, 502, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Okada, Y.; Hirokawa, N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 2005, 435, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, A.I.; Huang, B.Q.; Ward, C.J.; Gradilone, S.A.; Banales, J.M.; Masyuk, T.V.; Radtke, B.; Splinter, P.L.; LaRusso, N.F. Biliary exosomes influence cholangiocyte regulatory mechanisms and proliferation through interaction with primary cilia. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G990–G999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, M.C.; Manganelli, L.; Woollard, J.R.; Masyuk, A.I.; Masyuk, T.V.; Tammachote, R.; Huang, B.Q.; Leontovich, A.A.; Beito, T.G.; Madden, B.J.; et al. Characterization of PKD Protein-Positive Exosome-Like Vesicles. J. Am. Soc. Nephrol. 2009, 20, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [Green Version]
- Basten, S.G.; Giles, R.H. Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis. Cilia 2013, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Raphael, K.L.; Strait, K.A.; Stricklett, P.K.; Miller, R.L.; Nelson, R.D.; Piontek, K.B.; Germino, G.G.; Kohan, D.E. Inactivation of Pkd1 in principal cells causes a more severe cystic kidney disease than in intercalated cells. Kidney Int. 2009, 75, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Tian, X.; Igarashi, P.; Pazour, G.J.; Somlo, S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 2013, 45, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Barone, S.; Zahedi, K.; Brooks, M.; Henske, E.P.; Yang, Y.; Zhang, E.; Bissler, J.J.; Yu, J.J.; Soleimani, M. Kidney intercalated cells and the transcription factor FOXi1 drive cystogenesis in tuberous sclerosis complex. Proc. Natl. Acad. Sci. USA 2021, 118, e2020190118. [Google Scholar] [CrossRef]
- Bissler, J.J.; Zadjali, F.; Bridges, D.; Astrinidis, A.; Barone, S.; Yao, Y.; Redd, J.R.; Siroky, B.J.; Wang, Y.; Finley, J.T.; et al. Tuberous sclerosis complex exhibits a new renal cystogenic mechanism. Physiol. Rep. 2019, 7, e13983. [Google Scholar] [CrossRef]
- Zadjali, F.; Kumar, P.; Yao, Y.; Johnson, D.; Astrinidis, A.; Vogel, P.; Gross, K.W.; Bissler, J.J. Tuberous Sclerosis Complex Axis Controls Renal Extracellular Vesicle Production and Protein Content. Int. J. Mol. Sci. 2020, 21, 1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Zadjali, F.; Yao, Y.; Siroky, B.; Astrinidis, A.; Gross, K.W.; Bissler, J.J. Tsc Gene Locus Disruption and Differences in Renal Epithelial Extracellular Vesicles. Front. Physiol. 2021, 12, 630933. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Barr, M.M. Cell–cell communication via ciliary extracellular vesicles: Clues from model systems. Essays Biochem. 2018, 62, 205–213. [Google Scholar] [PubMed]
- Lin, F.; Hiesberger, T.; Cordes, K.; Sinclair, A.M.; Goldstein, L.S.B.; Somlo, S.; Igarashi, P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2003, 100, 5286–5291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Zadjali, F.; Yao, Y.; Johnson, D.; Siroky, B.; Astrinidis, A.; Vogel, P.; Gross, K.W.; Bissler, J.J. Tsc2 mutation induces renal tubular cell nonautonomous disease. Genes Dis. 2022, 9, 187–200. [Google Scholar] [CrossRef]
- Ding, H.; Li, L.X.; Harris, P.C.; Yang, J.; Li, X. Extracellular vesicles and exosomes generated from cystic renal epithelial cells promote cyst growth in autosomal dominant polycystic kidney disease. Nat. Commun. 2021, 12, 4548. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Olcese, J.M.; Meckes, D.G. Extraction of Extracellular Vesicles from Whole Tissue. J. Vis. Exp. 2019, e59143. [Google Scholar] [CrossRef]
- Cantagrel, V.; Silhavy, J.L.; Bielas, S.L.; Swistun, D.; Marsh, S.E.; Bertrand, J.Y.; Audollent, S.; Attié-Bitach, T.; Holden, K.R.; Dobyns, W.B.; et al. Mutations in the Cilia Gene ARL13B Lead to the Classical Form of Joubert Syndrome. Am. J. Hum. Genet. 2008, 83, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Siroky, B.J.; Kleene, N.K.; Kleene, S.J.; Varnell, C.D.; Comer, R.G.; Liu, J.; Lu, L.; Pachciarz, N.W.; Bissler, J.J.; Dixon, B.P. Primary cilia regulate the osmotic stress response of renal epithelial cells through TRPM3. Am. J. Physiol. Physiol. 2017, 312, F791–F805. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, A.; Takahashi, Y.; Chang, H.-Y.Y.; Wu, Y.-W.W.; Yamamoto, A.; Ishihama, Y.; Takakura, Y. Blood concentrations of small extracellular vesicles are determined by a balance between abundant secretion and rapid clearance. J. Extracell. Vesicles 2020, 9, 1696517. [Google Scholar] [CrossRef] [Green Version]
- Vidal, M. Exosomes: Revisiting their role as “garbage bags”. Traffic 2019, 20, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Seternes, T.; Sørensen, K.; Smedsrød, B. Scavenger endothelial cells of vertebrates: A nonperipheral leukocyte system for high-capacity elimination of waste macromolecules. Proc. Natl. Acad. Sci. USA 2002, 99, 7594–7597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandari, S.; Larsen, A.K.; McCourt, P.; Smedsrød, B.; Sørensen, K.K. The Scavenger Function of Liver Sinusoidal Endothelial Cells in Health and Disease. Front. Physiol. 2021, 12, 757469. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Jordan, V.; Blenkiron, C.; Chamley, L.W. Biodistribution of extracellular vesicles following administration into animals: A systematic review. J. Extracell. Vesicles 2021, 10, e12085. [Google Scholar] [CrossRef] [PubMed]
- Christ, L.; Raiborg, C.; Wenzel, E.M.; Campsteijn, C.; Stenmark, H. Cellular Functions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends Biochem. Sci. 2016, 42, 42–56. [Google Scholar] [CrossRef]
- Trajkovic, K. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 320, 179. [Google Scholar] [CrossRef]
- Theos, A.C.; Truschel, S.T.; Tenza, D.; Hurbain, I.; Harper, D.C.; Berson, J.F.; Thomas, P.C.; Raposo, G.; Marks, M.S. A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev. Cell 2006, 10, 343–354. [Google Scholar] [CrossRef] [Green Version]
- van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The Tetraspanin CD63 Regulates ESCRT-Independent and -Dependent Endosomal Sorting during Melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [Green Version]
- Siroky, B.J.; Yin, H.; Bissler, J.J. Clinical and Molecular Insights into Tuberous Sclerosis Complex Renal Disease. Pediatr. Nephrol. 2010, 26, 839–852. [Google Scholar] [CrossRef]
- Wang, J.; Nikonorova, I.A.; Silva, M.; Walsh, J.D.; Tilton, P.E.; Gu, A.; Akella, J.S.; Barr, M.M. Sensory cilia act as a specialized venue for regulated extracellular vesicle biogenesis and signaling. Curr. Biol. 2021, 31, 3943–3951.e3. [Google Scholar] [CrossRef]
- Wang, J.; Barr, M.M. Ciliary Extracellular Vesicles: Txt Msg Organelles. Cell. Mol. Neurobiol. 2016, 36, 449–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinas, R.Y.; Pearring, J.N.; Ding, J.-D.D.; Spencer, W.J.; Hao, Y.; Arshavsky, V.Y. Photoreceptor discs form through peripherindependent suppression of ciliary ectosome release. J. Cell Biol. 2017, 216, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phua, S.C.; Chiba, S.; Suzuki, M.; Su, E.; Roberson, E.C.; Pusapati, G.V.; Setou, M.; Rohatgi, R.; Reiter, J.F.; Ikegami, K.; et al. Dynamic Remodeling of Membrane Composition Drives Cell Cycle through Primary Cilia Excision. Cell 2017, 168, 264–279.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perut, F.; Roncuzzi, L.; Baldini, N. The Emerging Roles of Extracellular Vesicles in Osteosarcoma. Front. Oncol. 2019, 9, 1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofherr, A.; Busch, T.; Huber, N.; Nold, A.; Bohn, A.; Viau, A.; Bienaimé, F.; Kuehn, E.W.; Arnold, S.J.; Köttgen, M. Efficient genome editing of differentiated renal epithelial cells. Pflugers Arch. Eur. J. Physiol. 2017, 469, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Berbari, N.F.; Sharma, N.; Malarkey, E.B.; Pieczynski, J.N.; Boddu, R.; Gaertig, J.; Guay-Woodford, L.; Yoder, B.K. Microtubule modifications and stability are altered by cilia perturbation and in cystic kidney disease. Cytoskelet. 2013, 70, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Yoder, B.K.; Tousson, A.; Millican, L.; Wu, J.H.; Bugg, C.E.J.; Schafer, J.A.; Balkovetz, D.F. Polaris, a protein disrupted in orpk mutant mice, is required for assembly of renal cilium. Am. J. Physiol. Renal Physiol. 2002, 282, F541–F552. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Pkd1 | Pkd2 | Ratio |
|---|---|---|---|
| kuptake | 0.70 | 0.05 | 14 |
| Bmax | 0.5 | 0.7 | 0.71 |
| KM | 75 | 45 | 1.67 |
| kclearance | 0.0075 | 0.04 | 0.19 |
| half-life (h) | 92.4 | 17.3 | 5.34 |
| Parameter | Tsc1 | Tsc2 | Ratio |
|---|---|---|---|
| kuptake | 0.06 | 0.12 | 0.5 |
| Bmax | 0.12 | 0.25 | 0.48 |
| KM | 5 | 15 | 0.33 |
| kclearance | 0.15 | 0.05 | 3 |
| half-life (h) | 4.62 | 13.86 | 0.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, P.; Zadjali, F.; Yao, Y.; Köttgen, M.; Hofherr, A.; Gross, K.W.; Mehta, D.; Bissler, J.J. Single Gene Mutations in Pkd1 or Tsc2 Alter Extracellular Vesicle Production and Trafficking. Biology 2022, 11, 709. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11050709
Kumar P, Zadjali F, Yao Y, Köttgen M, Hofherr A, Gross KW, Mehta D, Bissler JJ. Single Gene Mutations in Pkd1 or Tsc2 Alter Extracellular Vesicle Production and Trafficking. Biology. 2022; 11(5):709. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11050709
Chicago/Turabian StyleKumar, Prashant, Fahad Zadjali, Ying Yao, Michael Köttgen, Alexis Hofherr, Kenneth W. Gross, Darshan Mehta, and John J. Bissler. 2022. "Single Gene Mutations in Pkd1 or Tsc2 Alter Extracellular Vesicle Production and Trafficking" Biology 11, no. 5: 709. https://0-doi-org.brum.beds.ac.uk/10.3390/biology11050709