
The Molecular Evolution, Structure, and Function of Coproporphyrinogen Oxidase and Protoporphyrinogen Oxidase in Prokaryotes



Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. List of Used Protein Sequences and Their Source
2.2. Multiple Protein Sequence Alignments
2.3. Reconstruction of Evolutionary Relationships
3. Results and Discussion
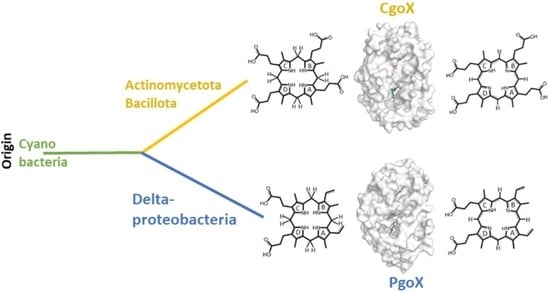
3.1. Phylogeny
3.2. Sequence Alignment and 3D Structures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rao, A.U.; Carta, L.K.; Lesuisse, E.; Hamza, I. Lack of Heme Synthesis in a Free-Living Eukaryote. Proc. Natl. Acad. Sci. USA 2005, 102, 4270–4275. [Google Scholar] [CrossRef] [PubMed]
- Sah, J.F.; Ito, H.; Kolli, B.K.; Peterson, D.A.; Sassa, S.; Chang, K.P. Genetic Rescue of Leishmania Deficiency in Porphyrin Biosynthesis Creates Mutants Suitable for Analysis of Cellular Events in Uroporphyria and for Photodynamic Therapy. J. Biol. Chem. 2002, 277, 14902–14909. [Google Scholar] [CrossRef] [PubMed]
- Baureder, M.; Hederstedt, L. Genes Important for Catalase Activity in Enterococcus Faecalis. PLoS ONE 2012, 7, e36725. [Google Scholar] [CrossRef] [PubMed]
- Dailey, H.A.; Dailey, T.A.; Gerdes, S.; Jahn, D.; Jahn, M.; O’Brian, M.R.; Warren, M.J. Prokaryotic Heme Biosynthesis: Multiple Pathways to a Common Essential Product. Microbiol. Mol. Biol. Rev. 2017, 81, e00048-16. [Google Scholar] [CrossRef] [PubMed]
- Dailey, H.A.; Gerdes, S.; Dailey, T.A.; Burch, J.S.; Phillips, J.D. Noncanonical Coproporphyrin-Dependent Bacterial Heme Biosynthesis Pathway That Does Not Use Protoporphyrin. Proc. Natl. Acad. Sci. USA 2015, 112, 2210–2215. [Google Scholar] [CrossRef]
- Dailey, H.A.; Medlock, A.E. A Primer on Heme Biosynthesis. Biol. Chem. 2022, 403, 985–1003. [Google Scholar] [CrossRef]
- Hamza, I.; Dailey, H.A. One Ring to Rule Them All: Trafficking of Heme and Heme Synthesis Intermediates in the Metazoans. Biochim. Biophys. Acta 2012, 1823, 1617–1632. [Google Scholar] [CrossRef]
- Adams, M.W.; Dailey, H.A.; DeLucas, L.J.; Luo, M.; Prestegard, J.H.; Rose, J.P.; Wang, B.C. The Southeast Collaboratory for Structural Genomics: A High-Throughput Gene to Structure factory. Acc. Chem. Res. 2003, 36, 191–198. [Google Scholar] [CrossRef]
- Hofbauer, S.; Helm, J.; Obinger, C.; Djinovic-Carugo, K.; Furtmüller, P.G. Crystal Structures and Calorimetry Reveal Catalytically Relevant Binding Mode of Coproporphyrin and Coproheme in Coproporphyrin Ferrochelatase. FEBS J. 2020, 287, 2779–2796. [Google Scholar] [CrossRef]
- Dali, A.; Gabler, T.; Sebastiani, F.; Destinger, A.; Furtmüller, P.G.; Pfanzagl, V.; Becucci, M.; Smulevich, G.; Hofbauer, S. Active Site Architecture of Coproporphyrin Ferrochelatase with Its Physiological Substrate Coproporphyrin III: Propionate Interactions and Porphyrin Core Deformation. Protein Sci. 2023, 32, e4534. [Google Scholar] [CrossRef]
- Gabler, T.; Sebastiani, F.; Helm, J.; Dali, A.; Obinger, C.; Furtmüller, P.G.; Smulevich, G.; Hofbauer, S. Substrate Specificity and Complex Stability of Coproporphyrin Ferrochelatase is Governed by Hydrogen-Bonding Interactions of the Four propionate groups. FEBS J. 2022, 289, 1680–1699. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, L.; Gabler, T.; Pfanzagl, V.; Michlits, H.; Furtmüller, P.G.; Obinger, C.; Hofbauer, S.; Smulevich, G. The Hydrogen Bonding Network of Coproheme in Coproheme Decarboxylase from Listeria Monocytogenes: Effect on Structure and Catalysis. J. Inorg. Biochem. 2019, 195, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Arnould, S.; Camadro, J.M. The Domain Structure of Protoporphyrinogen Oxidase, the Molecular Target of Diphenyl Ether-Type Herbicides. Proc. Natl. Acad. Sci. USA 1998, 95, 10553–10558. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Breithaupt, C.; Kiefersauer, R.; Freigang, J.; Huber, R.; Messerschmidt, A. Crystal Structure of Protoporphyrinogen IX Oxidase: A Key Enzyme in Haem and Chlorophyll Biosynthesis. EMBO J. 2004, 23, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An Improved General Amino Acid Replacement Matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Dailey, H.A.; Gerdes, S. HemQ: An Iron-Coproporphyrin Oxidative Decarboxylase for Protoheme Synthesis in Firmicutes and Actinobacteria. Arch. Biochem. Biophys. 2015, 574, 27–35. [Google Scholar] [CrossRef]
- Kobayashi, K.; Masuda, T.; Tajima, N.; Wada, H.; Sato, N. Molecular Phylogeny and Intricate Evolutionary History of the Three Isofunctional Enzymes Involved in the Oxidation of Protoporphyrinogen IX. Genome Biol. Evol. 2014, 6, 2141–2155. [Google Scholar] [CrossRef]
- Kohata, R.; Lim, H.; Kanamoto, Y.; Murakami, A.; Fujita, Y.; Tanaka, A.; Swingley, W.; Ito, H.; Tanaka, R. Heterologous complementation systems verify the mosaic distribution of three distinct protoporphyrinogen IX oxidase in the cyanobacterial phylum. J. Plant Res. 2023, 136, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Palenik, B.; Haselkorn, R. Multiple Evolutionary Origins of Prochlorophytes, the Chlorophyll b-Containing Prokaryotes. Nature 1992, 355, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Sessions, A.L.; Doughty, D.M.; Welander, P.V.; Summons, R.E.; Newman, D.K. The Continuing Puzzle of the Great Oxidation event. Curr. Biol. 2009, 19, R567–R574. [Google Scholar] [CrossRef] [PubMed]
- Corradi, H.R.; Corrigall, A.V.; Boix, E.; Mohan, C.G.; Sturrock, E.D.; Meissner, P.N.; Acharya, K.R. Crystal Structure of Protoporphyrinogen oxidase from Myxococcus xanthus and Its Complex with the Inhibitor Acifluorfen. J. Biol. Chem. 2006, 281, 38625–38633. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.H.; Tan, Y.; Wang, L.L.; Wang, Z.F.; Wang, B.F.; Wen, X.; Yang, G.F.; Xi, Z.; Shen, Y.Q. Structural Insight into Human Variegate Porphyria Disease. FASEB J. 2011, 25, 653–664. [Google Scholar] [CrossRef]
- Qin, X.H.; Sun, L.; Wen, X.; Yang, X.; Tan, Y.; Jin, H.; Cao, Q.Y.; Zhou, W.H.; Xi, Z.; Shen, Y.Q. Structural Insight into Unique Properties of Protoporphyrinogen Oxidase from Bacillus subtilis. J. Struct. Biol. 2010, 170, 76–82. [Google Scholar] [CrossRef]
- Wang, B.; Sun, L.; Wen, X.; Xi, Z. An Interaction Network in Bacillus subtilis Coproporphyrinogen Oxidase is Essential for the Oxidation of Protoporphyrinogen IX. Proteins 2023, 91, 1163–1172. [Google Scholar] [CrossRef]
- Sun, L.; Wen, X.; Tan, Y.; Li, H.Y.; Yang, X.; Zhao, Y.F.; Wang, B.F.; Cao, Q.Y.; Niu, C.W.; Xi, Z. Site-Directed Mutagenesis and Computational Study of the Y366 Active site in Bacillus subtilis Protoporphyrinogen Oxidase. Amino Acids 2009, 37, 523–530. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zámocký, M.; Hofbauer, S.; Gabler, T.; Furtmüller, P.G. The Molecular Evolution, Structure, and Function of Coproporphyrinogen Oxidase and Protoporphyrinogen Oxidase in Prokaryotes. Biology 2023, 12, 1527. https://0-doi-org.brum.beds.ac.uk/10.3390/biology12121527
Zámocký M, Hofbauer S, Gabler T, Furtmüller PG. The Molecular Evolution, Structure, and Function of Coproporphyrinogen Oxidase and Protoporphyrinogen Oxidase in Prokaryotes. Biology. 2023; 12(12):1527. https://0-doi-org.brum.beds.ac.uk/10.3390/biology12121527
Chicago/Turabian StyleZámocký, Marcel, Stefan Hofbauer, Thomas Gabler, and Paul G. Furtmüller. 2023. "The Molecular Evolution, Structure, and Function of Coproporphyrinogen Oxidase and Protoporphyrinogen Oxidase in Prokaryotes" Biology 12, no. 12: 1527. https://0-doi-org.brum.beds.ac.uk/10.3390/biology12121527