
Disease Transmission by Misfolded Prion-Protein Isoforms, Prion-Like Amyloids, Functional Amyloids and the Central Dogma

{kind=link}
Abstract
:1. Introduction

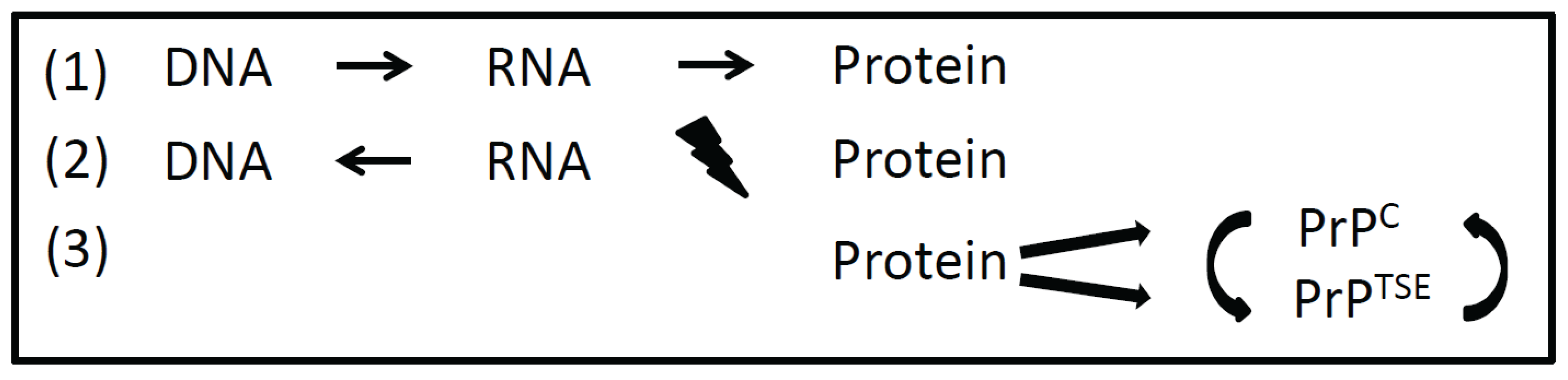
2. Expanding the Central Dogma
3. Prions as Information Carriers within and among Cells and Organisms
4. Prion-Like Amyloids and Functional Amyloids in Yeast and Bacteria
5. Conclusions
Conflicts of Interest
References
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, C.; Enari, M.; Klohn, P.C.; Rossi, D.; Flechsig, E. Molecular biology of prions. Acta Neurobiol. Exp. 2002, 62, 153–166. [Google Scholar]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Cohen, F.E.; Prusiner, S.B. Pathologic conformations of prion proteins. Annu. Rev. Biochem. 1998, 67, 793–819. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.W.; Dong, A.; Bhat, K.S.; Ernst, D.; Hayes, S.F.; Caughey, W.S. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 1991, 30, 7672–7680. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.D.; Matthews, S.J. New insight into the molecular control of bacterial functional amyloids. Front. Cell. Infect. Microbiol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Shewmaker, F.P.; Bateman, D.A.; Edskes, H.K.; Gorkovskiy, A.; Dayani, Y.; Bezsonov, E.E. Yeast prions: Structure, biology, and prion-handling systems. Microbiol. Mol. Biol. Rev. 2015, 79, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H. On protein synthesis. Symp. Soc. Exp. Biol. 1958, 12, 138–163. [Google Scholar]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Wang, D.W.; Wang, F.; Noble, G.P.; Ma, J.; Woods, V.L., Jr.; Li, S.; Supattapone, S. Cofactor molecules induce structural transformation during infectious prion formation. Structure 2013, 21, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, F. Prion disease and the “protein-only hypothesis”. Essays Biochem. 2014, 56, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Weber, P.; Sarafoff, N.; Beekes, M.; Giese, A.; Kretzschmar, H. Autocatalytic self-propagation of misfolded prion protein. Proc. Natl. Acad. Sci. USA 2004, 101, 12207–12211. [Google Scholar] [CrossRef] [PubMed]
- Daus, M.L.; Landmesser, H.; Schlosser, A.; Muller, P.; Herrmann, A.; Schneider, E. ATP induces conformational changes of periplasmic loop regions of the maltose atp-binding cassette transporter. J. Boil. Chem. 2006, 281, 3856–3865. [Google Scholar] [CrossRef] [PubMed]
- Daus, M.L.; Grote, M.; Muller, P.; Doebber, M.; Herrmann, A.; Steinhoff, H.J.; Dassa, E.; Schneider, E. ATP-driven malk dimer closure and reopening and conformational changes of the “EAA” motifs are crucial for function of the maltose ATP-binding cassette transporter (MalFGK2). J. Boil. Chem. 2007, 282, 22387–22396. [Google Scholar] [CrossRef] [PubMed]
- Takada, L.T.; Geschwind, M.D. Prion diseases. Sem. Neurol 2013, 33, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Daus, M.L.; Beekes, M. Chronic wasting disease: Fingerprinting the culprit in risk assessments. Prion 2012, 6, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.E.; Bartelt-Hunt, S.L.; Bartz, J.C. Occurrence, transmission, and zoonotic potential of chronic wasting disease. Emerg. Infect. Dis. 2012, 18, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Daus, M.L.; Breyer, J.; Wagenfuehr, K.; Wemheuer, W.M.; Thomzig, A.; Schulz-Schaeffer, W.J.; Beekes, M. Presence and seeding activity of pathological prion protein (PrP(TSE)) in skeletal muscles of white-tailed deer infected with chronic wasting disease. PLoS ONE 2011, 6, e18345. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, H.; Tatzelt, J. Prion disease: A tale of folds and strains. Brain Pathol. 2013, 23, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Requena, J.R.; Wille, H. The structure of the infectious prion protein: Experimental data and molecular models. Prion 2014, 8, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Daus, M.L. Techniques to elucidate the conformation of prions. World J. Biol. Chem. 2015, 6, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.L.; Kim, C.; Haldiman, T.; ElHag, M.; Mehndiratta, P.; Pichet, T.; Lissemore, F.; Shea, M.; Cohen, Y.; Chen, W.; et al. Rapidly progressive alzheimer's disease features distinct structures of amyloid-β. Brain 2015, 138, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Melki, R. Role of different alpha-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J. Parkinsons Dis. 2015, 5, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Simoneau, S.; Thomzig, A.; Ruchoux, M.M.; Vignier, N.; Daus, M.L.; Poleggi, A.; Lebon, P.; Freire, S.; Durand, V.; Graziano, S.; et al. Synthetic scrapie infectivity: Interaction between recombinant PrP and scrapie brain-derived RNA. Virulence 2015, 6, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J.; Lindquist, S. Prions as adaptive conduits of memory and inheritance. Nature reviews. Genetics 2005, 6, 435–450. [Google Scholar] [PubMed]
- Garrity, S.J.; Sivanathan, V.; Dong, J.; Lindquist, S.; Hochschild, A. Conversion of a yeast prion protein to an infectious form in bacteria. Proc. Natl. Acad. Sci. USA 2010, 107, 10596–10601. [Google Scholar] [CrossRef] [PubMed]
- Maclea, K.S.; Ross, E.D. Strategies for identifying new prions in yeast. Prion 2011, 5, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Cascarina, S.M.; Ross, E.D. Yeast prions and human prion-like proteins: Sequence features and prediction methods. Cell. Mol. Life Sci. 2014, 71, 2047–2063. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, Y.O. Amyloidogenic domains, prions and structural inheritance: Rudiments of early life or recent acquisition? Curr. Opin. Chem. Boil. 2004, 8, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.L. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold Des. 1998, 3, R9–R23. [Google Scholar] [CrossRef]
- Wang, X.; Chapman, M.R. Curli provide the template for understanding controlled amyloid propagation. Prion 2008, 2, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.Q.; Botyanszki, Z.; Tay, P.K.; Joshi, N.S. Programmable biofilm-based materials from engineered curli nanofibres. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.L.; Chapman, M.R. Curli biogenesis: Order out of disorder. Biochim. Biophys. Acta 2014, 1843, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Peggion, C.; Sorgato, M.C.; Bertoli, A. Prions and prion-like pathogens in neurodegenerative disorders. Pathogens 2014, 3, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.L.; Chorell, E.; Taylor, J.D.; Aden, J.; Gotheson, A.; Li, F.; Koch, M.; Sefer, L.; Matthews, S.J.; Wittung-Stafshede, P.; et al. The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol. Cell 2015, 57, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Requena, J.R. Prion-like diseases: Looking for their niche in the realm of infectious diseases. Virus Res. 2015, 207, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.; Ortiz, C.; Guzman, F.; Fernandez-Lafuente, R.; Torres, R. Antimicrobial peptides: Promising compounds against pathogenic microorganisms. Curr. Med. Chem. 2014, 21, 2299–2321. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daus, M.L. Disease Transmission by Misfolded Prion-Protein Isoforms, Prion-Like Amyloids, Functional Amyloids and the Central Dogma. Biology 2016, 5, 2. https://0-doi-org.brum.beds.ac.uk/10.3390/biology5010002
Daus ML. Disease Transmission by Misfolded Prion-Protein Isoforms, Prion-Like Amyloids, Functional Amyloids and the Central Dogma. Biology. 2016; 5(1):2. https://0-doi-org.brum.beds.ac.uk/10.3390/biology5010002
Chicago/Turabian StyleDaus, Martin L. 2016. "Disease Transmission by Misfolded Prion-Protein Isoforms, Prion-Like Amyloids, Functional Amyloids and the Central Dogma" Biology 5, no. 1: 2. https://0-doi-org.brum.beds.ac.uk/10.3390/biology5010002