
Structural Aspect of Hydroxyethyl-Starch–Anticancer-Drug-Conjugates as State-of-the-Art Drug Carriers


Abstract
:1. Introduction
2. Hydroxyethyl Starch (HES)
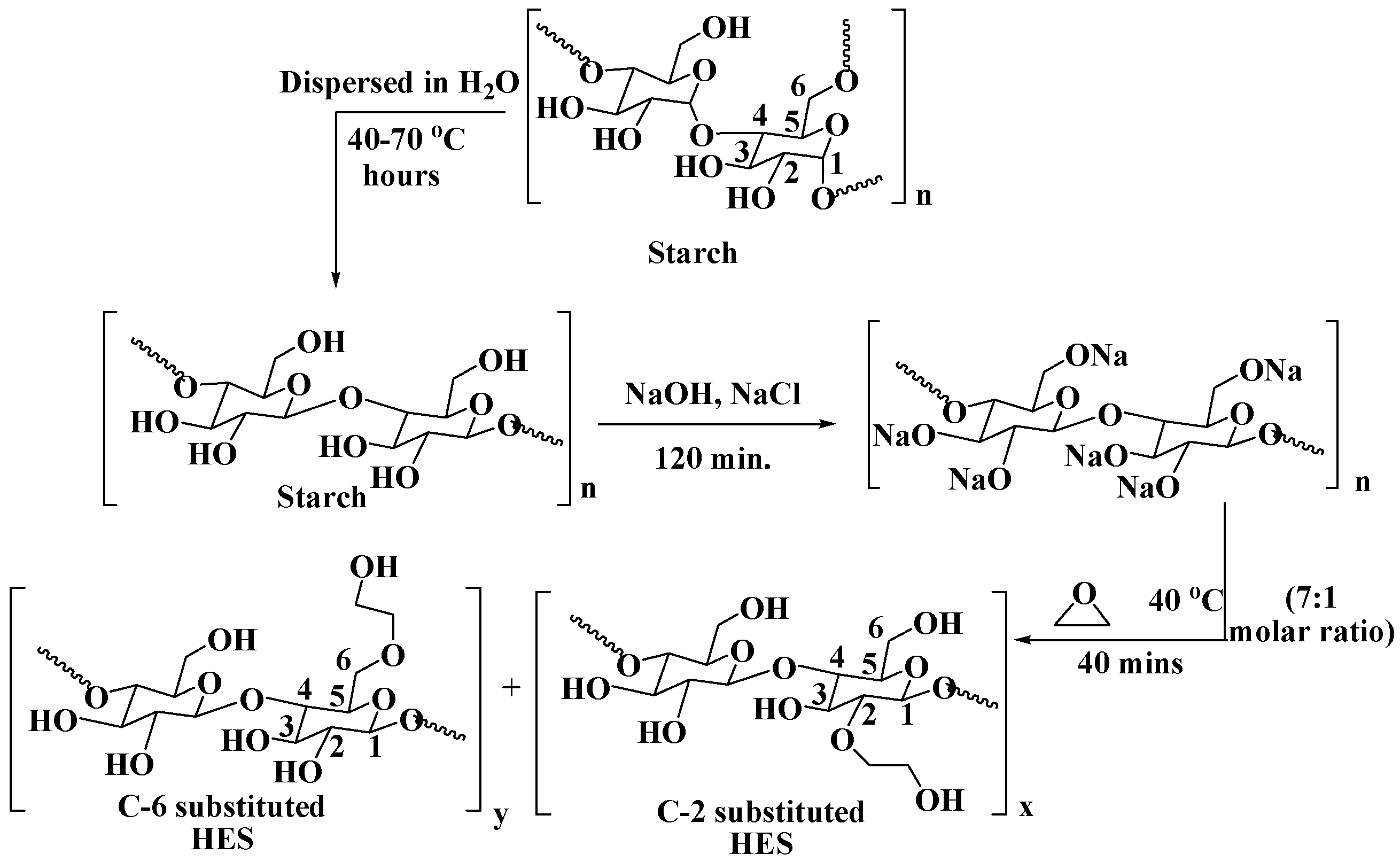
2.1. Synthesis
2.2. Synthesis of HES Based Polymers for Drug-delivery system
2.2.1. Self-Assemble
2.2.2. Layer-by-Layer Assembly
2.2.3. Nanoprecipitation
2.2.4. Graft Copolymerization
2.3. Commonly Used Anticancer Drugs
2.4. Drug-Delivery Systems (DDSs)
EPR Effect on DDS
2.5. Hydroxyethyl-Starch-Based Drug-Delivery System
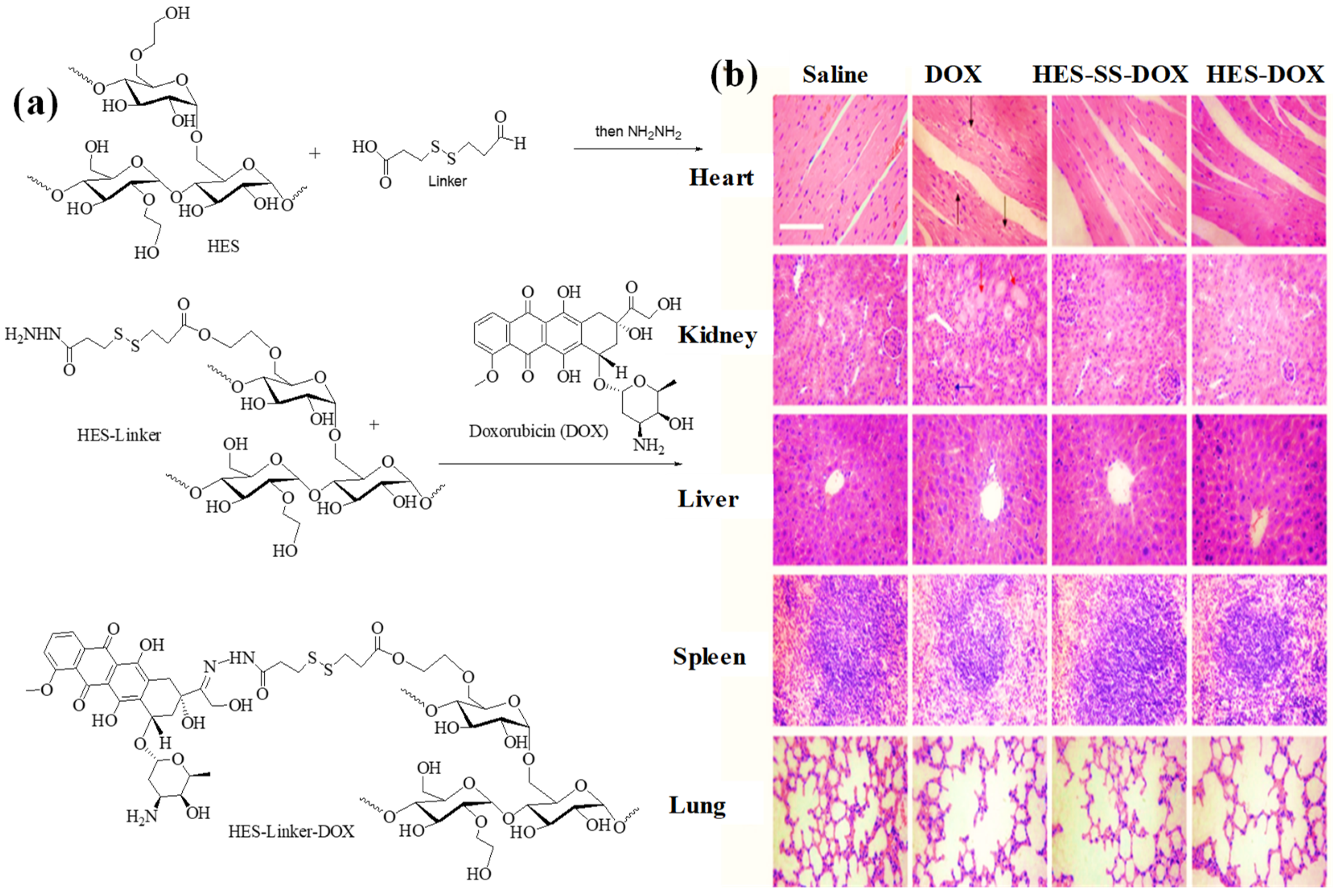
2.5.1. HES as DDS for Doxorubicin
2.5.2. HES as DDS for Methotrexate
2.5.3. HES as DDS for Hydroxychloroquine
2.5.4. HES as DDS for 5-Fluorouracil-1-acetic Acid
2.5.5. HES as DDS for 10-Hydroxy Camptothecin
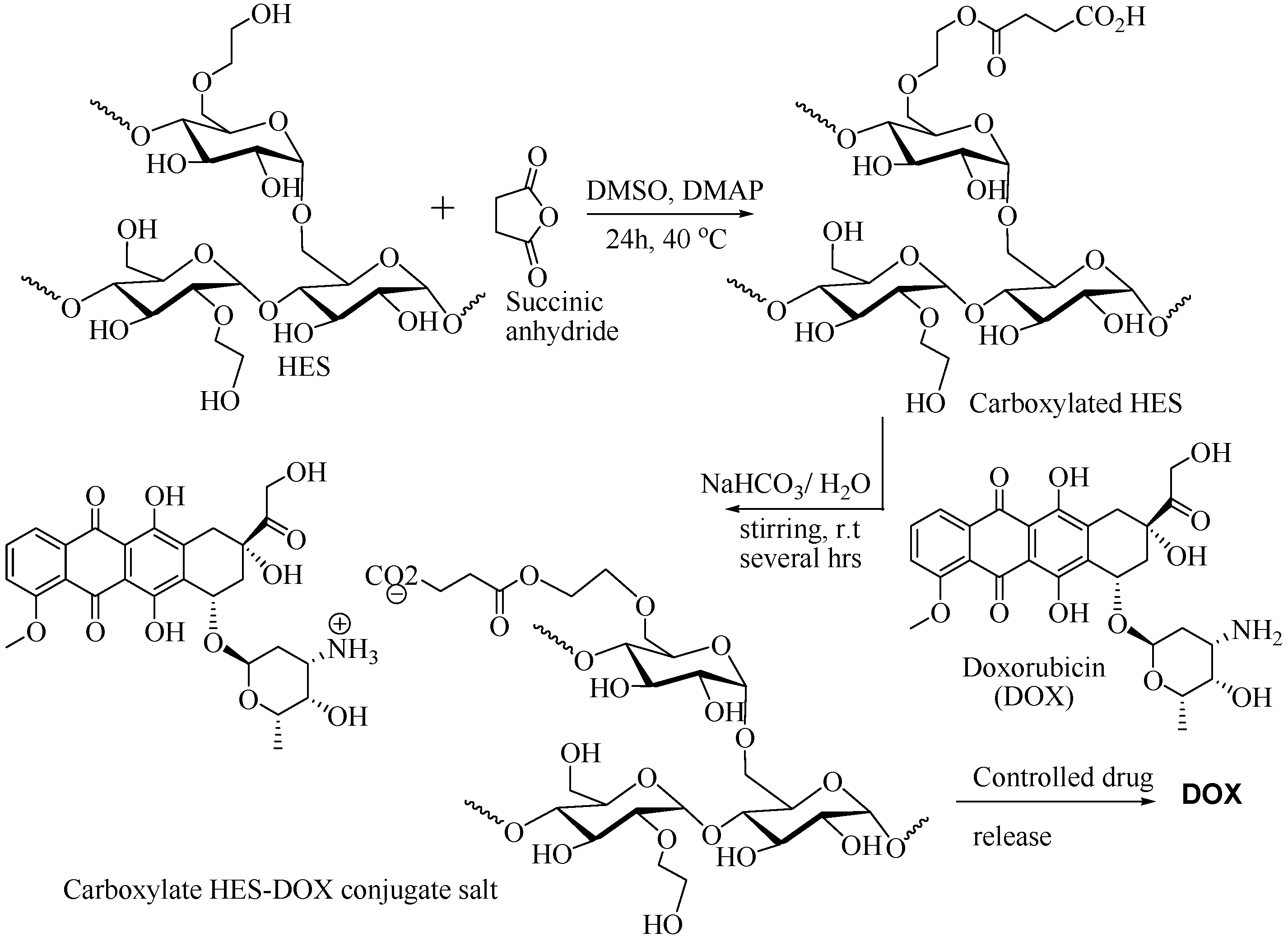
2.5.6. Carboxylated HES as DDS for Doxorubicin
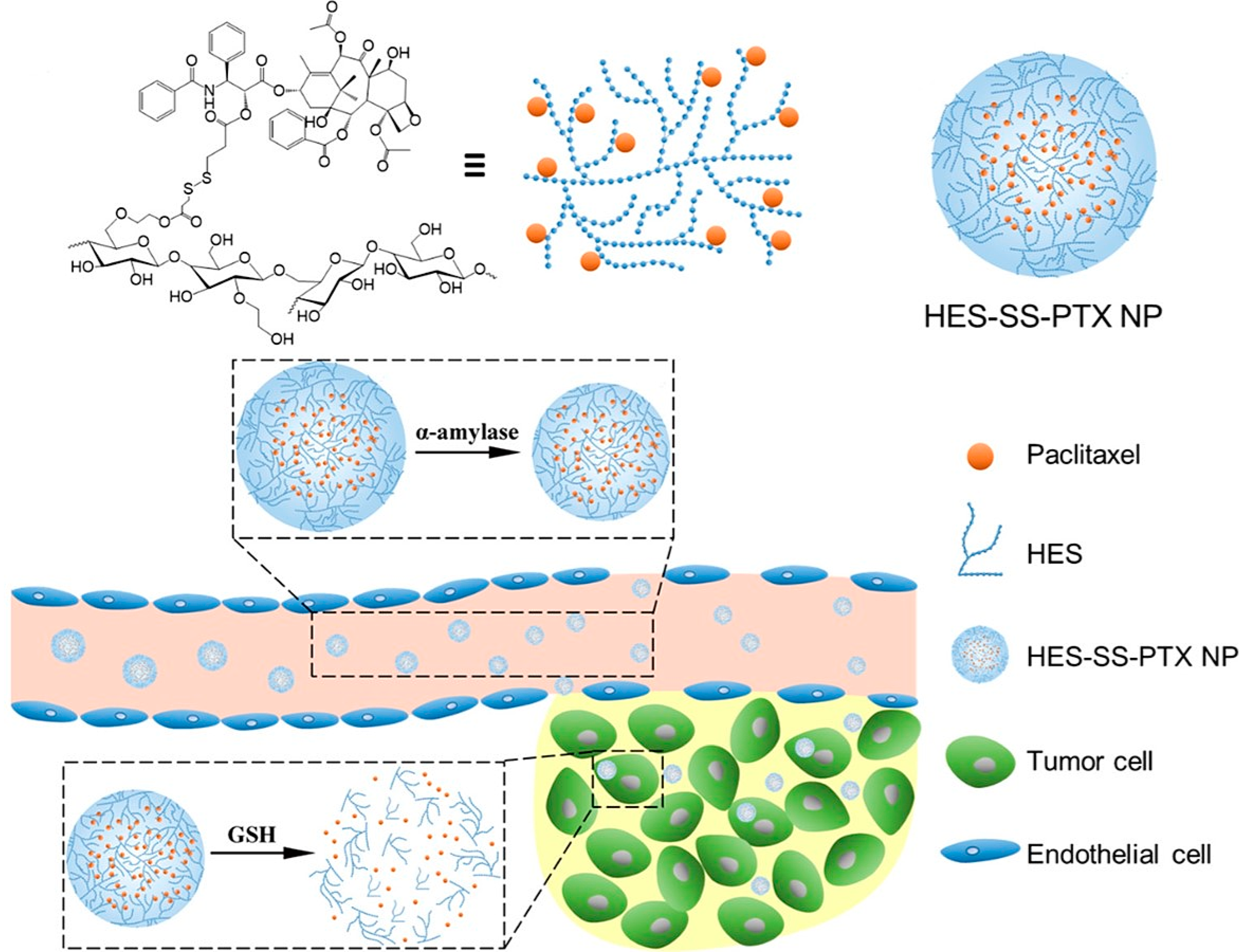
2.5.7. HES as DDS for Paclitaxel
2.5.8. HES-HEMA as DDS for Fluorescein Isothiocyanate
2.5.9. HES-PLGA as DDS
3. Limitations and Future Scope
- Although HES-based DDSs have shown promise in preclinical studies, there are still some challenges that need to be addressed before they can be translated into clinical applications.
- One limitation of HES-based DDS is associated with its immunogenicity. It might cause unknown or adverse immune response in some patients.
- The relatively short half-life of HES-DDS in the bloodstream may limit its effectiveness as a drug carrier.
- The variability in the properties of different HES formulations may affect their pharmacodynamic feasibility in the application of drug delivery.
- Future research could focus on improving the biocompatibility of HES-based DDS to minimize potential immune reactions.
- Efforts could also be made to optimize the properties of HES formulations to enhance their efficacy as drug carriers.
- Future research could involve by exploring the use of HES-based DDSs for targeted drug delivery to specific tissues or cells.
- The combination of HES with other natural or synthetic polymers can be investigated to develop more advanced delivery systems with improved properties.
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bertram, J.S. The molecular biology of cancer. Mol. Aspects Med. 2000, 21, 167–223. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA. Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobue, T. Association of Indoor Air Pollution and Lifestyle with Lung Cancer in Osaka, Japan. Int. J. Epidemiol. 1990, 19, S62–S66. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Pershagen, G. Cancer risk of air pollution: Epidemiological evidence. Environ. Health Perspect. 1994, 102, 187–192. [Google Scholar] [CrossRef]
- Ames, B.N.; Gold, L.S. Environmental pollution, pesticides, and the prevention of cancer: Misconceptions. FASEB J. 1997, 11, 1041–1052. [Google Scholar] [CrossRef] [Green Version]
- Drake, I.; Dias, J.A.; Teleka, S.; Stocks, T.; Orho-Melander, M. Lifestyle and cancer incidence and mortality risk depending on family history of cancer in two prospective cohorts. Int. J. Cancer 2019, 146, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Sriraman, S.K.; Aryasomayajula, B.; Torchilin, V.P. Barriers to drug delivery in solid tumors. Tissue Barriers 2014, 2, e29528. [Google Scholar] [CrossRef] [Green Version]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Bertino, J.R. Karnofsky memorial lecture. Ode to methotrexate. J. Clin. Oncol. 1993, 11, 5–14. [Google Scholar] [CrossRef]
- Kwon, G.S. Polymeric micelles for delivery of poorly water-soluble compounds. Crit. Rev. Ther. Drug Carr. Syst. 2003, 20, 357–403. [Google Scholar] [CrossRef]
- Cairns, R.; Papandreou, I.; Denko, N. Overcoming Physiologic Barriers to Cancer Treatment by Molecularly Targeting the Tumor Microenvironment. Mol. Cancer Res. 2006, 4, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Avdeef, A. Physicochemical Profiling (Solubility, Permeability and Charge State). Curr. Top. Med. Chem. 2001, 1, 277–351. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.J.A. and L.R. Tumor Physiology and Charge Dynamics of Anticancer Drugs: Implications for Camptothecin-based Drug Development. Curr. Med. Chem. 2011, 18, 1367–1372. [Google Scholar]
- Jacob, J.; Haponiuk, J.T.; Thomas, S.; Gopi, S. Biopolymer based nanomaterials in drug delivery systems: A review. Mater. Today Chem. 2018, 9, 43–55. [Google Scholar] [CrossRef]
- Debele, T.A.; Mekuria, S.L.; Tsai, H.-C. Polysaccharide based nanogels in the drug delivery system: Application as the carrier of pharmaceutical agents. Mater. Sci. Eng. C 2016, 68, 964–981. [Google Scholar] [CrossRef]
- Yu, C.; Liu, C.; Wang, S.; Li, Z.; Hu, H.; Wan, Y.; Yang, X. Hydroxyethyl Starch-Based Nanoparticles Featured with Redox-Sensitivity and Chemo-Photothermal Therapy for Synergized Tumor Eradication. Cancers 2019, 11, 207. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.; Opatz, T.; Landfester, K.; Wurm, F.R. Carbohydrate nanocarriers in biomedical applications: Functionalization and construction. Chem. Soc. Rev. 2015, 44, 8301–8325. [Google Scholar] [CrossRef] [Green Version]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chemie Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef]
- Wang, H.; Hu, H.; Yang, H.; Li, Z. Hydroxyethyl starch based smart nanomedicine. RSC Adv. 2021, 11, 3226–3240. [Google Scholar] [CrossRef]
- Tan, R.; Wan, Y.; Yang, X. Hydroxyethyl starch and its derivatives as nanocarriers for delivery of diagnostic and therapeutic agents towards cancers. Biomater. Transl. 2020, 1, 46. [Google Scholar] [PubMed]
- Devy, J.; Balasse, E.; Kaplan, H.; Madoulet, C.; Andry, M.-C. Hydroxyethylstarch microcapsules: A preliminary study for tumor immunotherapy application. Int. J. Pharm. 2006, 307, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Lemarchand, C.; Gref, R.; Couvreur, P. Polysaccharide-decorated nanoparticles. Eur. J. Pharm. Biopharm. 2004, 58, 327–341. [Google Scholar] [CrossRef]
- Noga, M.; Edinger, D.; Kläger, R.; Wegner, S.V.; Spatz, J.P.; Wagner, E.; Winter, G.; Besheer, A. The effect of molar mass and degree of hydroxyethylation on the controlled shielding and deshielding of hydroxyethyl starch-coated polyplexes. Biomaterials 2013, 34, 2530–2538. [Google Scholar] [CrossRef]
- Besheer, A.; Vogel, J.; Glanz, D.; Kressler, J.; Groth, T.; Mäder, K. Characterization of PLGA Nanospheres Stabilized with Amphiphilic Polymers: Hydrophobically Modified Hydroxyethyl Starch vs Pluronics. Mol. Pharm. 2009, 6, 407–415. [Google Scholar] [CrossRef]
- Perner, A.; Haase, N.; Guttormsen, A.B.; Tenhunen, J.; Klemenzson, G.; Åneman, A.; Madsen, K.R.; Møller, M.H.; Elkjær, J.M.; Poulsen, L.M.; et al. Hydroxyethyl Starch 130/0.42 versus Ringer’s Acetate in Severe Sepsis. N. Engl. J. Med. 2012, 367, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, E.L.; Duncan, R. Dextrin−Phospholipase A2: Synthesis and Evaluation as a Bioresponsive Anticancer Conjugate. Biomacromolecules 2009, 10, 1358–1364. [Google Scholar] [CrossRef]
- Marchant, R.E.; Yuan, S.; Szakalas-Gratzl, G. Interactions of plasma proteins with a novel polysaccharide surfactant physisorbed to polyethylene. J. Biomater. Sci. Polym. Ed. 1995, 6, 549–564. [Google Scholar] [CrossRef]
- Ruttmann, T.G.; James, M.F.; Aronson, I. In vivo investigation into the effects of haemodilution with hydroxyethyl starch (200/0.5) and normal saline on coagulation. Br. J. Anaesth. 1998, 80, 612–616. [Google Scholar] [CrossRef]
- de Jonge, E.; Levi, M.; Büller, H.; Berends, F.; Kesecioglu, J. Decreased circulating levels of von Willebrand factor after intravenous administration of a rapidly degradable hydroxyethyl starch (HES 200/0.5/6) in healthy human subjects. Intensive Care Med. 2001, 27, 1825–1829. [Google Scholar] [CrossRef]
- Isoda, S.; Izubuchi, R.; Yamazaki, I.; Nakayama, Y.; Yano, Y.; Masuda, M. Priming and replenishment in cardiopulmonary bypass with hydroxyethyl starch 130/0.4 decreases fluid overbalance without renal dysfunction or bleeding in adult valve surgery. Gen. Thorac. Cardiovasc. Surg. 2019, 67, 374–376. [Google Scholar] [CrossRef]
- Trentini, A.; Murganti, F.; Rosta, V.; Cervellati, C.; Manfrinato, C.M.; Spadaro, S.; Dallocchio, F.; Volta, A.C.; Bellini, T. Hydroxyethyl Starch 130/0.4 Binds to Neutrophils Impairing Their Chemotaxis through a Mac-1 Dependent Interaction. Int. J. Mol. Sci. 2019, 20, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.L.; Lautenschläger, I.; Zitta, K.; Hummitzsch, L.; Parczany, K.; Steinfath, M.; Weiler, N.; Albrecht, M. Effects of hydroxyethyl starch (HES 130/0.42) on endothelial and epithelial permeability in vitro. Toxicol. Vitr. 2019, 60, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Kolya, H.; Tripathy, T. Hydroxyethyl Starch-g-Poly-(N,N-dimethylacrylamide-co-acrylic acid): An efficient dye removing agent. Eur. Polym. J. 2013, 49, 4265–4275. [Google Scholar] [CrossRef]
- Jamnicki, M.; Zollinger, A.; Seifert, B.; Popovic, D.; Pasch, T.; Spahn, D.R. Compromised blood coagulation: An in vitro comparison of hydroxyethyl starch 130/0.4 and hydroxyethyl starch 200/0.5 using thrombelastography. Anesth. Analg. 1998, 87, 989–993. [Google Scholar]
- Langeron, O.; Doelberg, M.; Ang, E.-T.; Bonnet, F.; Capdevila, X.; Coriat, P. Voluven®, a lower substituted novel hydroxyethyl starch (HES 130/0.4), causes fewer effects on coagulation in major orthopedic surgery than HES 200/0.5. Anesth. Analg. 2001, 92, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Treib, J.; Haass, A.; Pindur, G.; Seyfert, U.T.; Treib, W.; Grauer, M.T.; Jung, F.; Wenzel, E.; Schimrigk, K. HES 200/0.5 Is not HES 200/0.5. Thromb. Haemost. 1995, 74, 1452–1456. [Google Scholar] [CrossRef]
- Standl, T.; Burmeister, M.-A.; Schroeder, F.; Currlin, E.; Esch, J.S.; Freitag, M.; Esch, J.S. Hydroxyethyl starch (HES) 130/0.4 provides larger and faster increases in tissue oxygen tension in comparison with prehemodilution values than HES 70/0.5 or HES 200/0.5 in volunteers undergoing acute normovolemic hemodilution. Anesth. Analg. 2003, 96, 936–943. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.; Tang, Q.; Xu, C.; Huang, Y.; Huang, D.; Luo, F.; Wu, Y.; Yan, F.; Weng, Z.; et al. Nano-micelles based on hydroxyethyl starch-curcumin conjugates for improved stability, antioxidant and anticancer activity of curcumin. Carbohydr. Polym. 2020, 228, 115398. [Google Scholar] [CrossRef]
- Li, G.; Li, Y.; Tang, Y.; Zhang, Y.; Zhang, Y.; Yin, T.; Xu, H.; Cai, C.; Tang, X. Hydroxyethyl starch conjugates for improving the stability, pharmacokinetic behavior and antitumor activity of 10-hydroxy camptothecin. Int. J. Pharm. 2014, 471, 234–244. [Google Scholar] [CrossRef]
- Treib, J.; Baron, J.-F.; Grauer, M.T.; Strauss, R.G. An international view of hydroxyethyl starches. Intensive Care Med. 1999, 25, 258–268. [Google Scholar] [CrossRef]
- Xiao, C.; Hu, H.; Yang, H.; Li, S.; Zhou, H.; Ruan, J.; Zhu, Y.; Yang, X.; Li, Z. Colloidal hydroxyethyl starch for tumor-targeted platinum delivery. Nanoscale Adv. 2019, 1, 1002–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baier, G.; Baumann, D.; Siebert, J.M.; Musyanovych, A.; Mailänder, V.; Landfester, K. Suppressing Unspecific Cell Uptake for Targeted Delivery Using Hydroxyethyl Starch Nanocapsules. Biomacromolecules 2012, 13, 2704–2715. [Google Scholar] [CrossRef]
- Xu, C.; Chen, S.; Chen, C.; Ming, Y.; Du, J.; Mu, J.; Luo, F.; Huang, D.; Wang, N.; Lin, Z.; et al. Colon-targeted oral nanoparticles based on ROS-scavenging hydroxyethyl starch-curcumin conjugates for efficient inflammatory bowel disease therapy. Int. J. Pharm. 2022, 623, 121884. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, J.; Dong, C.; Zhu, L.; Zhou, L.; Zhu, K. Hydroxyethyl Starch Improves the Prognosis of Rats with Traumatic Shock via Activation of the ERK Signaling Pathway in Lymphocytes. Comput. Math. Methods Med. 2022, 2022, 5262189. [Google Scholar] [CrossRef] [PubMed]
- El-Hinnawy, S.I.; Fahmy, A.; El-Saied, H.M.; El-Shirbeeny, A.F.; El-Sahy, K.M. Preparation and evaluation of hydroxyethyl starch. Starch-Stärke 1982, 34, 65–68. [Google Scholar] [CrossRef]
- El-Sahy, K.M.; Ahmed, S.H.; Attia, R.M.; Fahmy, A.H. Studies on the kinetics of amyloglucosidase as affected by native and derivatized corn starch. Food Chem. 1984, 15, 45–50. [Google Scholar] [CrossRef]
- Öztürk, Y.S.; Dolaz, M. Synthesis and Characterization of Hydroxyethyl Starch from Chips Wastes Under Microwave Irradiation. J. Polym. Environ. 2021, 29, 948–957. [Google Scholar] [CrossRef]
- Glover, P.A.; Rudloff, E.; Kirby, R. Hydroxyethyl starch: A review of pharmacokinetics, pharmacodynamics, current products, and potential clinical risks, benefits, and use. J. Vet. Emerg. Crit. Care 2014, 24, 642–661. [Google Scholar] [CrossRef]
- Xia, Y.; Yin, Y.; Lu, Y.; McLellan, J. Template-Assisted Self-Assembly of Spherical Colloids into Complex and Controllable Structures. Adv. Funct. Mater. 2003, 13, 907–918. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, Z.; Yang, B. Self-assembly of photonic crystals from polymer colloids. Curr. Opin. Colloid Interface Sci. 2009, 14, 103–114. [Google Scholar] [CrossRef]
- Poon, Z.; Lee, J.B.; Morton, S.W.; Hammond, P.T. Controlling in Vivo Stability and Biodistribution in Electrostatically Assembled Nanoparticles for Systemic Delivery. Nano Lett. 2011, 11, 2096–2103. [Google Scholar] [CrossRef] [PubMed]
- Mizrahy, S.; Peer, D. Polysaccharides as building blocks for nanotherapeutics. Chem. Soc. Rev. 2012, 41, 2623–2640. [Google Scholar] [CrossRef] [PubMed]
- Fessi, H.; Puisieux, F.; Devissaguet, J.P.; Ammoury, N.; Benita, S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 1989, 55, R1–R4. [Google Scholar] [CrossRef]
- Roy, D.; Semsarilar, M.; Guthrie, J.T.; Perrier, S. Cellulose modification by polymer grafting: A review. Chem. Soc. Rev. 2009, 38, 2046–2064. [Google Scholar] [CrossRef]
- Oh, J.K.; Lee, D.I.; Park, J.M. Biopolymer-based microgels/nanogels for drug delivery applications. Prog. Polym. Sci. 2009, 34, 1261–1282. [Google Scholar] [CrossRef]
- Wei, H.; Cheng, S.-X.; Zhang, X.-Z.; Zhuo, R.-X. Thermo-sensitive polymeric micelles based on poly(N-isopropylacrylamide) as drug carriers. Prog. Polym. Sci. 2009, 34, 893–910. [Google Scholar] [CrossRef]
- Ju, B.; Yan, D.; Zhang, S. Micelles self-assembled from thermoresponsive 2-hydroxy-3-butoxypropyl starches for drug delivery. Carbohydr. Polym. 2012, 87, 1404–1409. [Google Scholar] [CrossRef]
- Yang, J.; Gao, C.; Lü, S.; Zhang, X.; Yu, C.; Liu, M. Physicochemical characterization of amphiphilic nanoparticles based on the novel starch–deoxycholic acid conjugates and self-aggregates. Carbohydr. Polym. 2014, 102, 838–845. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, X.; Shu, X.; Shen, Z.; Sun, R.-C. Self-Assembly and Paclitaxel Loading Capacity of Cellulose-graft-poly(lactide) Nanomicelles. J. Agric. Food Chem. 2012, 60, 3900–3908. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Ma, Y.; Chen, F.; Zhao, G. Synthesis, Characterization, and Aqueous Self-Assembly of Octenylsuccinate Oat β-Glucan. J. Agric. Food Chem. 2013, 61, 12683–12691. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, M.; Guo, Y.; Wang, X.; Zhang, L.; Zhou, J.; Li, H.; Shi, Q. Self-assembly of cationic amphiphilic cellulose-g-poly (p-dioxanone) copolymers. Carbohydr. Polym. 2019, 204, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.J.; Ding, C.; Qu, X.; Yang, Z.; Uchegbu, I.F.; Tetley, L.; Cheng, W.P. The effect of polymer architecture on the nano self-assemblies based on novel comb-shaped amphiphilic poly(allylamine). Colloid Polym. Sci. 2008, 286, 1511–1526. [Google Scholar] [CrossRef]
- Besheer, A.; Hause, G.; Kressler, J.; Mäder, K. Hydrophobically Modified Hydroxyethyl Starch: Synthesis, Characterization, and Aqueous Self-Assembly into Nano-Sized Polymeric Micelles and Vesicles. Biomacromolecules 2007, 8, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.; Jones, S.A.; Lvov, Y.M. Biomedical applications of electrostatic layer-by-layer nano-assembly of polymers, enzymes, and nanoparticles. Cell Biochem. Biophys. 2003, 39, 23. [Google Scholar] [CrossRef] [PubMed]
- Yadav, K.S.; Sawant, K.K. Modified Nanoprecipitation Method for Preparation of Cytarabine-Loaded PLGA Nanoparticles. AAPS PharmSciTech 2010, 11, 1456–1465. [Google Scholar] [CrossRef] [Green Version]
- Quintanar-Guerrero, D.; Allémann, E.; Fessi, H.; Doelker, E. Preparation Techniques and Mechanisms of Formation of Biodegradable Nanoparticles from Preformed Polymers. Drug Dev. Ind. Pharm. 1998, 24, 1113–1128. [Google Scholar] [CrossRef]
- Šebenik, A. Living free-radical block copolymerization using thio-iniferters. Prog. Polym. Sci. 1998, 23, 875–917. [Google Scholar] [CrossRef]
- Jenkins, D.W.; Hudson, S.M. Review of Vinyl Graft Copolymerization Featuring Recent Advances toward Controlled Radical-Based Reactions and Illustrated with Chitin/Chitosan Trunk Polymers. Chem. Rev. 2001, 101, 3245–3274. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living Radical Polymerization by the RAFT Process. Aust. J. Chem. 2005, 58, 379–410. [Google Scholar] [CrossRef]
- Lizundia, E.; Meaurio, E.; Vilas, J.L. Chapter 3-Grafting of Cellulose Nanocrystals. In Multifunctional Polymeric Nanocomposites Based on Cellulosic Reinforcements; Puglia, D., Fortunati, E., Kenny, J.M., Eds.; William Andrew Publishing: Norwich, NY, USA, 2016; pp. 61–113. ISBN 978-0-323-44248-0. [Google Scholar]
- Borch, R.F.; Bernstein, M.D.; Durst, H.D. Cyanohydridoborate anion as a selective reducing agent. J. Am. Chem. Soc. 1971, 93, 2897–2904. [Google Scholar] [CrossRef]
- Noga, M.; Edinger, D.; Rödl, W.; Wagner, E.; Winter, G.; Besheer, A. Controlled shielding and deshielding of gene delivery polyplexes using hydroxyethyl starch (HES) and alpha-amylase. J. Control. Release 2012, 159, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Z.; Huang, L. pH and thermo dual-responsive starch-g-P(DEAEMA-co-PEGMA): Synthesis via SET-LRP, self-assembly and drug release behaviors. React. Funct. Polym. 2019, 141, 165–171. [Google Scholar] [CrossRef]
- Yu, C.; Zhou, Q.; Xiao, F.; Li, Y.; Hu, H.; Wan, Y.; Li, Z.; Yang, X. Enhancing Doxorubicin Delivery toward Tumor by Hydroxyethyl Starch-g-Polylactide Partner Nanocarriers. ACS Appl. Mater. Interfaces 2017, 9, 10481–10493. [Google Scholar] [CrossRef]
- Singh, Y.; Palombo, M.; Sinko, P.J. Recent trends in targeted anticancer prodrug and conjugate design. Curr. Med. Chem. 2008, 15, 1802–1826. [Google Scholar] [CrossRef] [Green Version]
- Khandare, J.; Minko, T. Polymer–drug conjugates: Progress in polymeric prodrugs. Prog. Polym. Sci. 2006, 31, 359–397. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs1. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef]
- Kwon, I.K.; Lee, S.C.; Han, B.; Park, K. Analysis on the current status of targeted drug delivery to tumors. J. Control. Release 2012, 164, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Leporatti, S. Thinking about Enhanced Permeability and Retention Effect (EPR). J. Pers. Med. 2022, 12, 1259. [Google Scholar] [CrossRef]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Park, K. Questions on the role of the EPR effect in tumor targeting. J. Control. Release 2013, 172, 391. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Kopečková, P.; Minko, T.; Rubinstein, A.; Kopeček, J. Design of a multivalent galactoside ligand for selective targeting of HPMA copolymer–doxorubicin conjugates to human colon cancer cells. Eur. J. Cancer 2004, 40, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Hu, H.; Wan, J.; Li, Y.; Wu, Y.; Tang, Y.; Xiao, C.; Xu, H.; Yang, X.; Li, Z. Hydroxyethyl starch stabilized polydopamine nanoparticles for cancer chemotherapy. Chem. Eng. J. 2018, 349, 129–145. [Google Scholar] [CrossRef]
- Li, D.; Ding, J.; Zhuang, X.; Chen, L.; Chen, X. Drug binding rate regulates the properties of polysaccharide prodrugs. J. Mater. Chem. B 2016, 4, 5167–5177. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Li, Y.; Zhou, Q.; Ao, Y.; Yu, C.; Wan, Y.; Xu, H.; Li, Z.; Yang, X. Redox-Sensitive Hydroxyethyl Starch–Doxorubicin Conjugate for Tumor Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2016, 8, 30833–30844. [Google Scholar] [CrossRef]
- Hu, H.; Xiao, C.; Wu, H.; Li, Y.; Zhou, Q.; Tang, Y.; Yu, C.; Yang, X.; Li, Z. Nanocolloidosomes with Selective Drug Release for Active Tumor-Targeted Imaging-Guided Photothermal/Chemo Combination Therapy. ACS Appl. Mater. Interfaces 2017, 9, 42225–42238. [Google Scholar] [CrossRef]
- Zhao, K.; Li, D.; Xu, W.; Ding, J.; Jiang, W.; Li, M.; Wang, C.; Chen, X. Targeted hydroxyethyl starch prodrug for inhibiting the growth and metastasis of prostate cancer. Biomaterials 2017, 116, 82–94. [Google Scholar] [CrossRef]
- Jiang, G.; Qiu, W.; DeLuca, P.P. Preparation and in Vitro/in Vivo Evaluation of Insulin-Loaded Poly(Acryloyl-Hydroxyethyl Starch)-PLGA Composite Microspheres. Pharm. Res. 2003, 20, 452–459. [Google Scholar] [CrossRef]
- Goszczyński, T.M.; Filip-Psurska, B.; Kempińska, K.; Wietrzyk, J.; Boratyński, J. Hydroxyethyl starch as an effective methotrexate carrier in anticancer therapy. Pharmacol. Res. Perspect. 2014, 2, e00047. [Google Scholar] [CrossRef]
- Li, G.; Zhao, M.; Zhao, L. Lysine-mediated hydroxyethyl starch-10-hydroxy camptothecin micelles for the treatment of liver cancer. Drug Deliv. 2020, 27, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, R.V.; Inamdar, S.Z.; Das, K.K.; Biradar, M.S. 7-Polysaccharide-based stimuli-sensitive graft copolymers for drug delivery. In Polysaccharide Carriers for Drug Delivery; Maiti, S., Jana, S., Eds.; Woodhead Publishing: Cambridge, UK, 2019; pp. 155–177. ISBN 978-0-08-102553-6. [Google Scholar]
- Mosbah, I.B.; Franco-Gou, R.; Abdennebi, H.B.; Hernandez, R.; Escolar, G.; Saidane, D.; Rosello-Catafau, J.; Peralta, C. Effects of Polyethylene Glycol and Hydroxyethyl Starch in University of Wisconsin Preservation Solution on Human Red Blood Cell Aggregation and Viscosity. Transplant. Proc. 2006, 38, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yang, X.; Xu, H.; Pan, K.; Yang, Y. Novel nanomicelles originating from hydroxyethyl starch-g-polylactide and their release behavior of docetaxel modulated by the PLA chain length. Eur. Polym. J. 2013, 49, 3522–3529. [Google Scholar] [CrossRef]
- Kolya, H.; Sasmal, D.; Tripathy, T. Novel Biodegradable Flocculating Agents Based on Grafted Starch Family for the Industrial Effluent Treatment. J. Polym. Environ. 2017, 25, 408–418. [Google Scholar] [CrossRef]
- Kulicke, W.-M.; Heinze, T. Improvements in Polysaccharides for use as Blood Plasma Expanders. Macromol. Symp. 2005, 231, 47–59. [Google Scholar] [CrossRef]
- Ickx, B.E.; Bepperling, F.; Melot, C.; Schulman, C.; Van der Linden, P.J. Plasma substitution effects of a new hydroxyethyl starch HES 130/0.4 compared with HES 200/0.5 during and after extended acute normovolaemic haemodilution. Br. J. Anaesth. 2003, 91, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Shi, B.; Xu, W.; Ding, J.; Yang, Y.; Liu, H.; Zhuang, X.; Chen, X. Reduction-responsive polypeptide nanogel delivers antitumor drug for improved efficacy and safety. Acta Biomater. 2015, 27, 179–193. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Dammer, E.B.; Ren, R.-J.; Wang, G. The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Sleightholm, R.; Yang, B.; Yu, F.; Xie, Y.; Oupický, D. Chloroquine-Modified Hydroxyethyl Starch as a Polymeric Drug for Cancer Therapy. Biomacromolecules 2017, 18, 2247–2257. [Google Scholar] [CrossRef]
- Wang, H.; Yu, J.; Lu, X.; He, X. Nanoparticle systems reduce systemic toxicity in cancer treatment. Nanomedicine 2015, 11, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Wang, P.; Miao, Y.; He, H.; Tang, X. A novel 5-fluorouracil prodrug using hydroxyethyl starch as a macromolecular carrier for sustained release. Carbohydr. Polym. 2012, 87, 2642–2647. [Google Scholar] [CrossRef]
- Gan, B.K.; Rullah, K.; Yong, C.Y.; Ho, K.L.; Omar, A.R.; Alitheen, N.B.; Tan, W.S. Targeted delivery of 5-fluorouracil-1-acetic acid (5-FA) to cancer cells overexpressing epithelial growth factor receptor (EGFR) using virus-like nanoparticles. Sci. Rep. 2020, 10, 16867. [Google Scholar] [CrossRef] [PubMed]
- Fugit, K.D.; Anderson, B.D. The role of pH and ring-opening hydrolysis kinetics on liposomal release of topotecan. J. Control. Release 2014, 174, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Cai, C.; Qi, Y.; Tang, X. Hydroxyethyl starch–10-hydroxy camptothecin conjugate: Synthesis, pharmacokinetics, cytotoxicity and pharmacodynamics research. Drug Deliv. 2016, 23, 277–284. [Google Scholar] [CrossRef]
- Paleos, C.M.; Sideratou, Z.; Theodossiou, T.A.; Tsiourvas, D. Carboxylated Hydroxyethyl Starch: A novel Polysaccharide for the Delivery of Doxorubicin. Chem. Biol. Drug Des. 2015, 85, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, H.; Zhou, Q.; Ao, Y.; Xiao, C.; Wan, J.; Wan, Y.; Xu, H.; Li, Z.; Yang, X. α-Amylase- and Redox-Responsive Nanoparticles for Tumor-Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2017, 9, 19215–19230. [Google Scholar] [CrossRef]
- Miller, K.; Eldar-Boock, A.; Polyak, D.; Segal, E.; Benayoun, L.; Shaked, Y.; Satchi-Fainaro, R. Antiangiogenic Antitumor Activity of HPMA Copolymer–Paclitaxel–Alendronate Conjugate on Breast Cancer Bone Metastasis Mouse Model. Mol. Pharm. 2011, 8, 1052–1062. [Google Scholar] [CrossRef]
- Harling, S.; Schwoerer, A.; Scheibe, K.; Daniels, R.; Menzel, H. A new hydrogel drug delivery system based on Hydroxyethylstarch derivatives. J. Microencapsul. 2010, 27, 400–408. [Google Scholar] [CrossRef]
- Wöhl-Bruhn, S.; Bertz, A.; Harling, S.; Menzel, H.; Bunjes, H. Hydroxyethyl starch-based polymers for the controlled release of biomacromolecules from hydrogel microspheres. Eur. J. Pharm. Biopharm. 2012, 81, 573–581. [Google Scholar] [CrossRef]
- Bertz, A.; Wöhl-Bruhn, S.; Miethe, S.; Tiersch, B.; Koetz, J.; Hust, M.; Bunjes, H.; Menzel, H. Encapsulation of proteins in hydrogel carrier systems for controlled drug delivery: Influence of network structure and drug size on release rate. J. Biotechnol. 2013, 163, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Woo, B.H.; Jiang, G.; Jo, Y.W.; DeLuca, P.P. Preparation and Characterization of a Composite PLGA and Poly(Acryloyl Hydroxyethyl Starch) Microsphere System for Protein Delivery. Pharm. Res. 2001, 18, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medicines and Therapy | Applications | Working Group Conclusions | |
|---|---|---|---|
| 1. | Azacitidine | Acute myeloid leukemia | Not support |
| 2. | Cancer medicines for children up to 12 years old, EMLc: carboplatin, cisplatin, cyclophosphamide, vinblastine and vincristine | Low-grade glioma | Supports |
| 3. | CAR-T cell therapy | Acute lymphoblastic leukemia | Supports |
| 4. | Cyclin-dependent kinase (CDK) 4/6 inhibitors | Metastatic breast cancer | Not support |
| 5. | Daratumumab | Multiple myeloma | Not support |
| 6. | Doxorubicin | Rhabdomyosarcoma | Supports |
| 7. | Enzalutamide | Metastatic castration-resistant prostate cancer | Supports |
| 8. | Everolimus | Subependymal giant cell astrocytoma | Supports |
| 9. | Fulvestrant | Metastatic breast cancer | Not support |
| 10. | Ibrutinib | chronic lymphocytic leukemia with 17p deletion | supports |
| 11. | Osimertinib | Lung cancer | Not support |
| 12. | Pembrolizumab | Locally advanced and metastatic non-small cell lung cancer | Supports |
| 13. | Pertuzumab | HER2+ metastatic breast cancer | Supports |
| 14. | Rasburicase | Tumor lysis syndrome | Supports |
| 15. | Tislelizumab | Hodgkin lymphoma | Not support |
| 16. | Tislelizumab | Urothelial carcinoma | Not support |
| 17. | Tyrosine kinase inhibitors | Ph+ acute lymphoblastic leukemia | Supports |
| 18. | Vinorelbine | Rhabdomyosarcoma | Supports |
| 19. | Zanubrutinib | chronic lymphocytic leukemia, small lymphocytic lymphoma | Not support |
| 20. | Zanubrutinib | Mantle cell lymphoma | Not support |
| Sl No | Biopolymers/Biopolymer-Based Polymers | Conjugated with Drug | Utilize for | IC50 (µg/mL) (Cell Line) | References |
|---|---|---|---|---|---|
| 1. | HES-polydopamine | Doxorubicin | Cancer chemotherapy | 0.89 (HepG2) | [85] |
| 2. | HES-Doxorubicin | Doxorubicin | Chemotherapy of malignancy | 7.12 (HepG2) | [86] |
| 3. | HES-SS-Doxorubicin | Doxorubicin | Safe cancer chemotherapy | 1.97 (HepG2) | [87] |
| 4. | HES-polycaprolactone-Doxorubicin-indocyanine green | Doxorubicin and indocyanine green mixture | Liver cancer | - | [88] |
| 5. | LHRH-conjugated HES-doxorubicin | Doxorubicin | Clinical chemotherapy of metastatic prostate cancer | 0.79 (RM-1) | [89] |
| 6. | HES-coated polydopamine nanoparticles | Doxorubicin | Antitumor drug for cancer chemotherapy | 0.46 (HepG2) | [85] |
| 7. | HES-g-Polylactide | Doxorubicin | Clinical cancer chemotherapy | - | [90] |
| 8. | HES-MTX | Methotrexate | Clinical cancer chemotherapy | 106 (MV4-11) | [91] |
| 9. | HES-10-HCPT-SS-Lysine | 10-Hydroxy camptothecin (10-HCPT) | Liver cancer chemotherapy | 9.9 (HepG2) | [92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandra, K.; Dutta, S.; Kolya, H.; Kang, C.-W. Structural Aspect of Hydroxyethyl-Starch–Anticancer-Drug-Conjugates as State-of-the-Art Drug Carriers. Sci. Pharm. 2023, 91, 32. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm91030032
Chandra K, Dutta S, Kolya H, Kang C-W. Structural Aspect of Hydroxyethyl-Starch–Anticancer-Drug-Conjugates as State-of-the-Art Drug Carriers. Scientia Pharmaceutica. 2023; 91(3):32. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm91030032
Chicago/Turabian StyleChandra, Koushik, Sansa Dutta, Haradhan Kolya, and Chun-Won Kang. 2023. "Structural Aspect of Hydroxyethyl-Starch–Anticancer-Drug-Conjugates as State-of-the-Art Drug Carriers" Scientia Pharmaceutica 91, no. 3: 32. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm91030032