
Deletion of Kvβ2 (AKR6) Attenuates Isoproterenol Induced Cardiac Injury with Links to Solute Carrier Transporter SLC41a3 and Circadian Clock Genes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
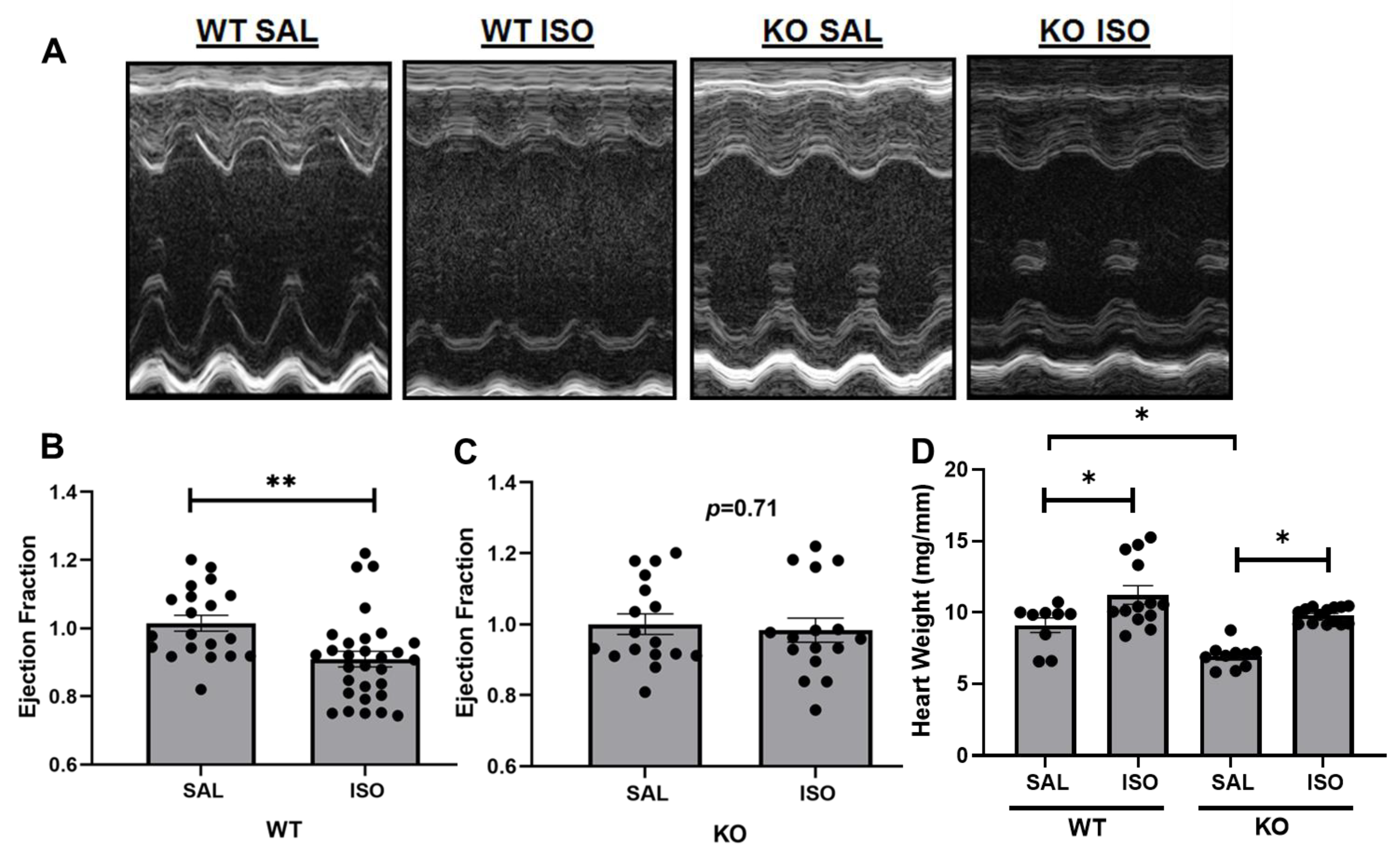
2.1. Echocardiographic Assessment Demonstrates a Differential Change in Cardiac Injury in Kvβ2 KO Mice by Isoproterenol Infusion
2.2. Kvβ2 Deletion Results in Significant Prolongation in QTc While Attenuated during Isoproterenol-Induced Cardiac Injury
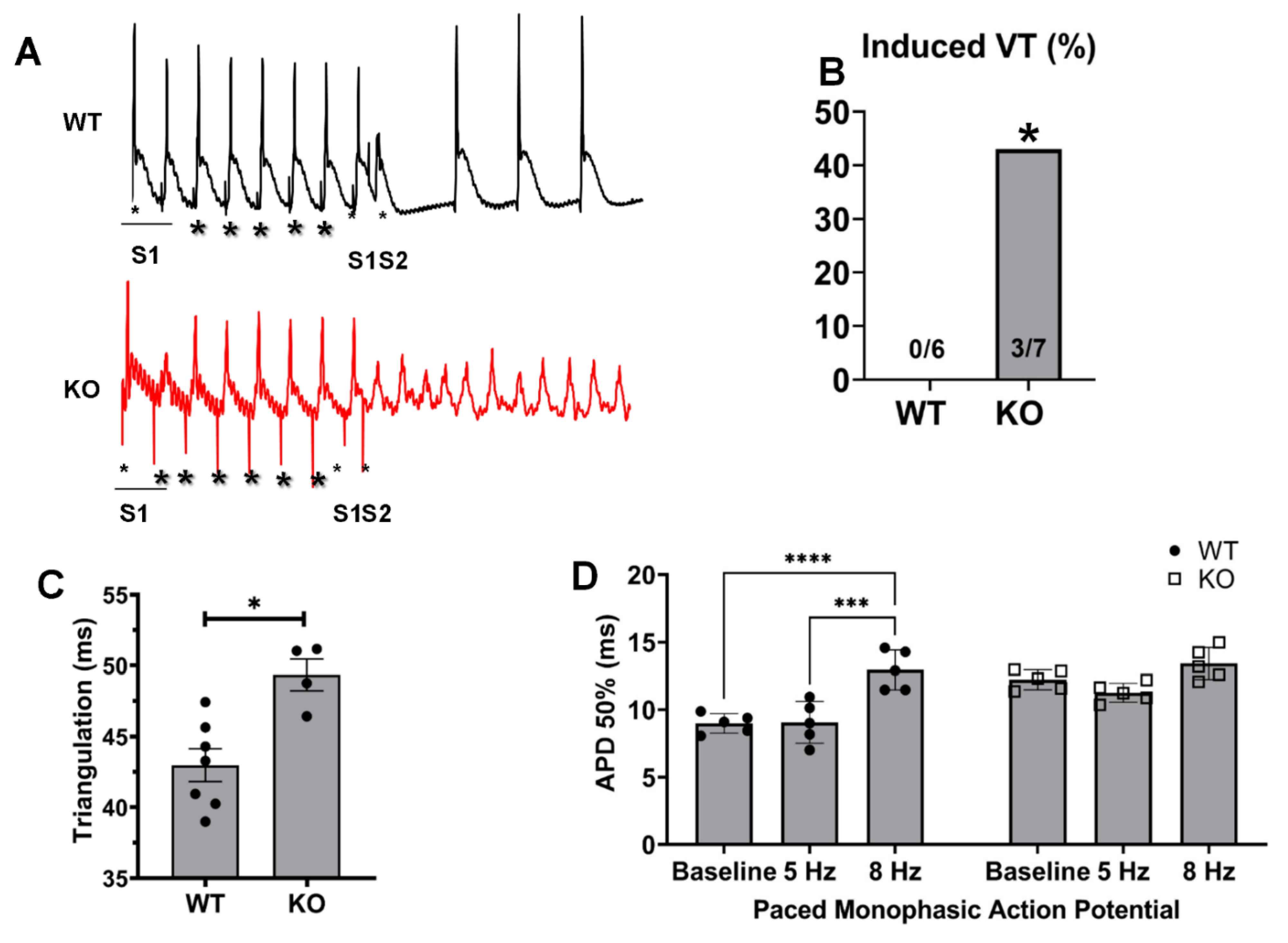
2.3. Changes in Monophasic Action Potentials in ISO-Infused Hearts
2.4. Kvβ2 Deletion Results in Significant Induction of Ventricular Tachycardias and Increased APDs
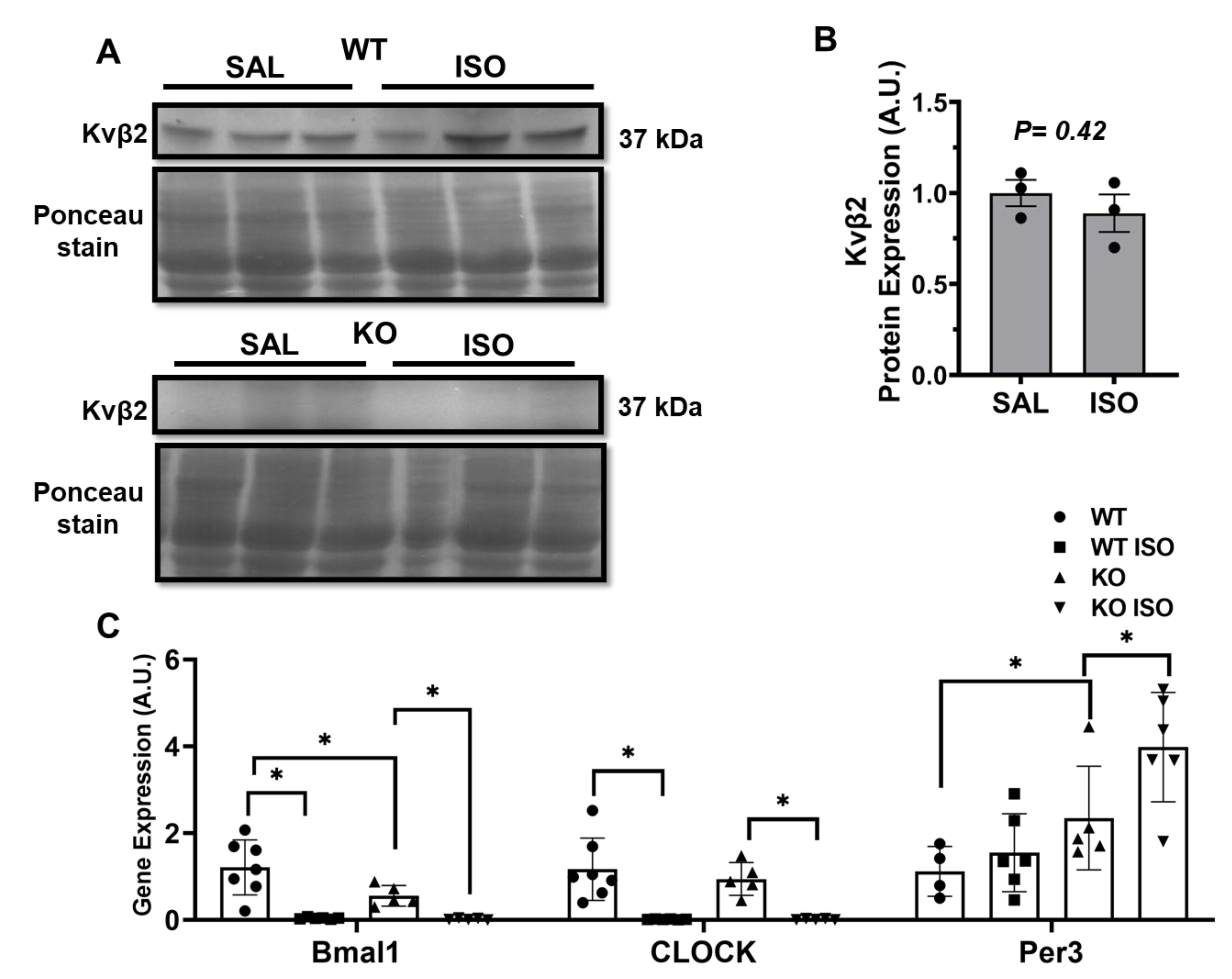
2.5. Microarray Analysis Identifies Novel Genes Altered in Kvβ2 KO Mice
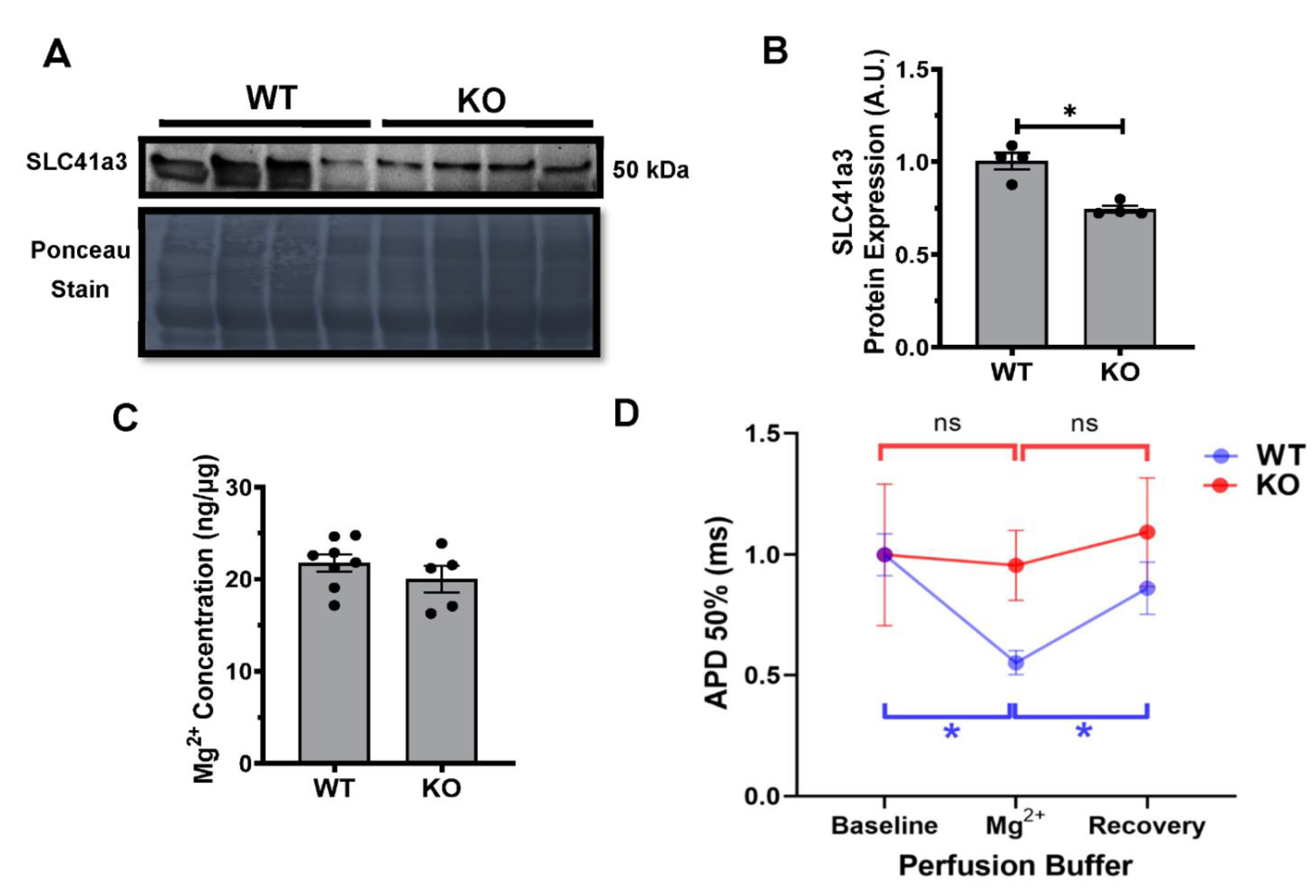
2.6. Kvβ2 KO Results in Significant Decrease in SLC41a3 Expression
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Mouse Model of Cardiac Hypertrophy
4.3. Echocardiography
4.4. Electrocardiography
4.5. Monophasic Action Potentials
4.6. Microarray
4.7. Quantitative Real-Time-PCR
4.8. Western Blots
4.9. Serum Magnesium (Mg2+) Concentrations
4.10. Microarray Data Analysis
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barski, O.A.; Tipparaju, S.M.; Bhatnagar, A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab. Rev. 2008, 40, 553–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilfoil, P.J.; Tipparaju, S.M.; Barski, O.A.; Bhatnagar, A. Regulation of ion channels by pyridine nucleotides. Circ. Res. 2013, 112, 721–741. [Google Scholar] [CrossRef] [Green Version]
- Tur, J.; Badole, S.L.; Cheng, F.; Das, A.; Kukreja, R.C.; Tipparaju, S.M. Corticosteroids and aldose reductase inhibitor Epalrestat modulates cardiac action potential via Kvβ1.1 (AKR6A8) subunit of voltage-gated potassium channel. Mol. Cell. Biochem. 2017, 436, 71–78. [Google Scholar] [CrossRef]
- Tipparaju, S.M.; Barski, O.A.; Srivastava, S.; Bhatnagar, A. Catalytic mechanism and substrate specificity of the beta-subunit of the voltage-gated potassium channel. Biochemistry 2008, 47, 8840–8854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkowski, J.J.; Murphy, G.G. Deletion of the Mouse Homolog of KCNAB2, a Gene Linked to Monosomy 1p36, Results in Associative Memory Impairments and Amygdala Hyperexcitability. J. Neurosci. 2011, 31, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Gajecka, M.; Mackay, K.L.; Shaffer, L.G. Monosomy 1p36 deletion syndrome. Am. J. Med Genet. 2007, 145C, 346–356. [Google Scholar] [CrossRef]
- Portero, V.; Scouarnec, S.L.; Es-Salah-Lamoureux, Z.; Burel, S.; Gourraud, J.B.; Bonnaud, S.; Lindenbaum, P.; Simonet, F.; Violleau, J.; Baron, E.; et al. Dysfunction of the Voltage&Gated K+ Channel β2 Subunit in a Familial Case of Brugada Syndrome. J. Am. Heart Assoc. 2016, 5, e003122. [Google Scholar] [CrossRef] [Green Version]
- Kilfoil, P.J.; Chapalamadugu, K.C.; Hu, X.; Zhang, D.; Raucci, F.J., Jr.; Tur, J.; Brittian, K.R.; Jones, S.P.; Bhatnagar, A.; Tipparaju, S.M.; et al. Metabolic regulation of Kv channels and cardiac repolarization by Kvβ2 subunits. J. Mol. Cell. Cardiol. 2019, 137, 93–106. [Google Scholar] [CrossRef]
- Wang, Y.; Mo, X.; Ping, C.; Huang, Q.; Zhang, H.; Xie, C.; Zhong, B.; Li, D.; Yao, J. Site-specific contacts enable distinct modes of TRPV1 regulation by the potassium channel Kvβ1 subunit. J. Biol. Chem. 2020, 295, 17337–17348. [Google Scholar] [CrossRef] [PubMed]
- Tur, J.; Chapalamadugu, K.C.; Katnik, C.; Cuevas, J.; Bhatnagar, A.; Tipparaju, S.M. Kvβ1.1 (AKR6A8) senses pyridine nucleotide changes in the mouse heart and modulates cardiac electrical activity. Am. J. Physiology. Heart Circ. Physiol. 2017, 312, H571–H583. [Google Scholar] [CrossRef]
- Pongs, O.; Schwarz, J.R. Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 2010, 90, 755–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barski, O.A.; Tipparaju, S.M.; Bhatnagar, A. Kinetics of nucleotide binding to the beta-subunit (AKR6A2) of the voltage-gated potassium (Kv) channel. Chem. Biol. Interact. 2009, 178, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinemann, S.H.; Rettig, J.; Wunder, F.; Pongs, O. Molecular and functional characterization of a rat brain Kv beta 3 potassium channel subunit. Febs Lett. 1995, 377, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Tipparaju, S.M.; Liu, S.Q.; Barski, O.A.; Bhatnagar, A. NADPH binding to beta-subunit regulates inactivation of voltage-gated K(+) channels. Biochem. Biophys. Res. Commun. 2007, 359, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ussher, J.R.; Jaswal, J.S.; Lopaschuk, G.D. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ. Res. 2012, 111, 628–641. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.; Hsu, C.P.; Sadoshima, J. Regulation of cell survival and death by pyridine nucleotides. Circ. Res. 2012, 111, 611–627. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Chun, Y.S.; Kim, M.S.; Park, Y.C.; Kwak, S.J.; Park, S.C. Metabolic modulation of cellular redox potential can improve cardiac recovery from ischemia-reperfusion injury. Int. J. Cardiol. 1998, 65, 139–147. [Google Scholar] [CrossRef]
- Pillai, J.B.; Gupta, M.; Rajamohan, S.B.; Lang, R.; Raman, J.; Gupta, M.P. Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1545–H1553. [Google Scholar] [CrossRef]
- Ceconi, C.; Bernocchi, P.; Boraso, A.; Cargnoni, A.; Pepi, P.; Curello, S.; Ferrari, R. New insights on myocardial pyridine nucleotides and thiol redox state in ischemia and reperfusion damage. Cardiovasc. Res. 2000, 47, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.J.; Honoré, E. Molecular physiology of oxygen-sensitive potassium channels. Eur. Respir. J. 2001, 18, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peri, R.; Wible, B.A.; Brown, A.M. Mutations in the Kv beta 2 binding site for NADPH and their effects on Kv1.4. J. Biol. Chem. 2001, 276, 738–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berul, C.I.; Sweeten, T.L.; Dubin, A.M.; Shah, M.J.; Vetter, V.L. Use of the rate-corrected JT interval for prediction of repolarization abnormalities in children. Am. J. Cardiol. 1994, 74, 1254–1257. [Google Scholar] [CrossRef]
- Frommeyer, G.; Eckardt, L. Drug-induced proarrhythmia: Risk factors and electrophysiological mechanisms. Nat. Rev. Cardiol. 2016, 13, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Osadchii, O.E. Role of abnormal repolarization in the mechanism of cardiac arrhythmia. Acta Physiol. 2017, 220, 1–71. [Google Scholar] [CrossRef] [Green Version]
- Fernández, J.C. New Methodologies for the Development and Validation of Electrophysiological Models. PhD Thesis, Universidad de Zaragoza, Zaragoza, Spain, 2019. [Google Scholar]
- Rabinovich-Nikitin, I.; Lieberman, B.; Martino, T.A.; Kirshenbaum, L.A. Circadian-Regulated Cell Death in Cardiovascular Diseases. Circulation 2019, 139, 965–980. [Google Scholar] [CrossRef]
- Kukreja, R.C. Myriad roles of voltage-activated potassium channel subunit Kvβ1.1 in the heart. Am. J. Physiology. Heart Circ. Physiol. 2017, 312, H546–H548. [Google Scholar] [CrossRef] [Green Version]
- Tatarkova, Z.; de Baaij, J.H.F.; Grendar, M.; Aschenbach, J.R.; Racay, P.; Bos, C.; Sponder, G.; Hoenderop, J.G.J.; Röntgen, M.; Turcanova Koprusakova, M.; et al. Dietary Mg2+ Intake and the Na+/Mg2+ Exchanger SLC41A1 Influence Components of Mitochondrial Energetics in Murine Cardiomyocytes. Int. J. Mol. Sci. 2020, 21, 8221. [Google Scholar] [CrossRef]
- Yu, N.; Jiang, J.; Yu, Y.; Li, H.; Huang, X.; Ma, Y.; Zhang, L.; Zou, J.; Zhang, B.; Chen, S.; et al. SLC41A1 knockdown inhibits angiotensin II-induced cardiac fibrosis by preventing Mg(2+) efflux and Ca(2+) signaling in cardiac fibroblasts. Arch. Biochem. Biophys. 2014, 564, 74–82. [Google Scholar] [CrossRef]
- Lamothe, S.M.; Sharmin, N.; Silver, G.; Satou, M.; Hao, Y.; Tateno, T.; Baronas, V.A.; Kurata, H.T. Control of Slc7a5 sensitivity by the voltage-sensing domain of Kv1 channels. eLife 2020, 9. [Google Scholar] [CrossRef]
- Redwood, S.R.; Taggart, P.I.; Sutton, P.M.; Bygrave, A.; Bashir, Y.; Purkayastha, D.D.; Camm, A.J.; Treasure, T. Effect of magnesium on the monophasic action potential during early ischemia in the in vivo human heart. J. Am. Coll Cardiol. 1996, 28, 1765–1769. [Google Scholar] [CrossRef] [Green Version]
- Hannu, J. Parikka, L.K.T. Acute Effects of Intravenous Magnesium on Ventricular Refractoriness and Monophasic Action Potential Duration in Humans. Scand. Cardiovasc. J. 1999, 33, 300–305. [Google Scholar] [CrossRef]
- Zhang, S.; Sawanobori, T.; Adaniya, H.; Hirano, Y.; Hiraoka, M. Dual effects of external magnesium on action potential duration in guinea pig ventricular myocytes. Am. J. Physiol. 1995, 268, H2321–H2328. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tur, J.; Chapalamadagu, K.C.; Manickam, R.; Cheng, F.; Tipparaju, S.M. Deletion of Kvβ2 (AKR6) Attenuates Isoproterenol Induced Cardiac Injury with Links to Solute Carrier Transporter SLC41a3 and Circadian Clock Genes. Metabolites 2021, 11, 201. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11040201
Tur J, Chapalamadagu KC, Manickam R, Cheng F, Tipparaju SM. Deletion of Kvβ2 (AKR6) Attenuates Isoproterenol Induced Cardiac Injury with Links to Solute Carrier Transporter SLC41a3 and Circadian Clock Genes. Metabolites. 2021; 11(4):201. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11040201
Chicago/Turabian StyleTur, Jared, Kalyan C. Chapalamadagu, Ravikumar Manickam, Feng Cheng, and Srinivas M. Tipparaju. 2021. "Deletion of Kvβ2 (AKR6) Attenuates Isoproterenol Induced Cardiac Injury with Links to Solute Carrier Transporter SLC41a3 and Circadian Clock Genes" Metabolites 11, no. 4: 201. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11040201