
Influence of Extraction Solvent on Nontargeted Metabolomics Analysis of Enrichment Reactor Cultures Performing Enhanced Biological Phosphorus Removal (EBPR)

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. General Profiles of LC-MS Features in Lab-Scale EBPR Bioreactor Microbial Communities
2.2. Extraction Capacity
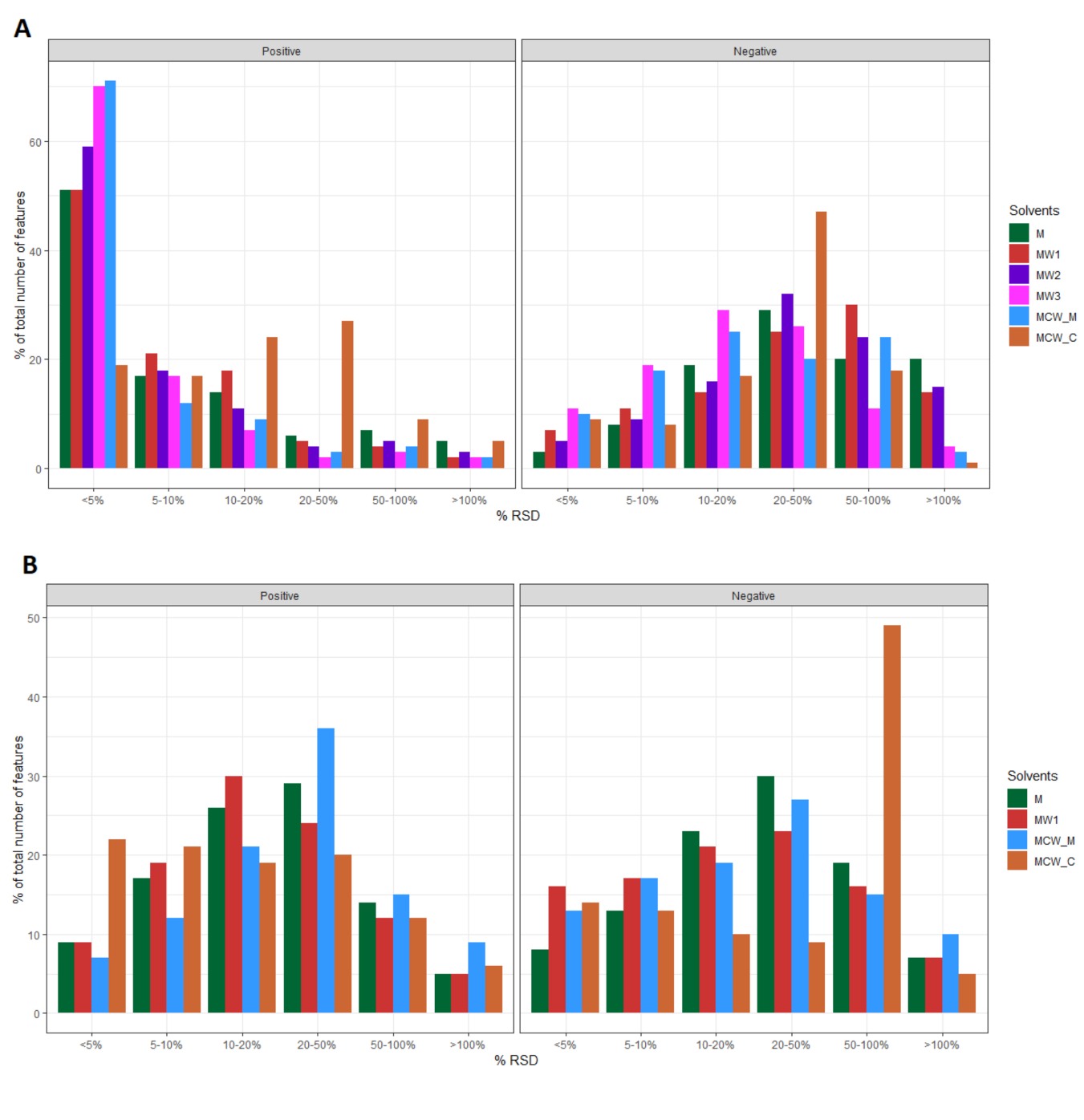
2.3. Evaluation of Method Repeatability
2.4. Biological Level Interpretation and Differentiation of Physiological States
3. Discussion
4. Materials and Methods
4.1. Operation of the Lab-Scale SBR Enrichment Bioreactors
4.2. Experimental Design, Sample Collection and Extraction Procedures
4.2.1. Methanol and Methanol: Water Extraction
4.2.2. Methanol: Chloroform: Water Extraction
4.3. UPLC-MS Analysis and Data Processing
4.4. Metabolite Analysis Using UPLC-MS
4.5. Lipid Profiling Using UPLC-MS
4.6. Data Processing
4.7. Statistical Analysis
16S-SSU-rRNA Amplicon Sequencing Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kido Soule, M.C.; Longnecker, K.; Johnson, W.M.; Kujawinski, E.B. Environmental metabolomics: Analytical strategies. Mar. Chem. 2015, 177, 374–387. [Google Scholar] [CrossRef] [Green Version]
- Peters, D.L.; Wang, W.-J.; Zhang, X.; Ning, Z.; Mayne, J.; Figys, D. Metaproteomic and metabolomic approaches for characterising the gut microbiome. Proteomics 2019, 19, 1800363. [Google Scholar] [CrossRef]
- Muller, E.E.L.; Pinel, N.; Laczny, C.C.; Hoopmann, M.R.; Narayanasamy, S.; Lebrun, L.A.; Roume, H. Community-integrated omics links dominance of a microbial generalist to fine-tuned resource usage. Nat. Commun. 2014, 5, 5603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peisl, B.Y.L.; Schymanski, E.L.; Wilmes, P. Dark matter in host-microbiome metabolomics: Tackling the unknowns—A review. Anal. Chim. Acta 2018, 1037, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.T.; Aksenov, A.A.; Nothias, L.F.; Foulds, J.R.; Quinn, R.A.; Badri, M.H.; Swenson, T.L.; Van Goethem, M.W.; Northen, T.R.; Vazquez-Baeza, Y.; et al. Learning representations of microbe-metabolite interactions. Nat. Methods 2019, 16, 1306–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louca, S.; Doebeli, M. Reaction-centric modeling of microbial ecosystems. Ecol. Model 2016, 335, 74–86. [Google Scholar] [CrossRef]
- Li, M.-J.; Qian, W.-J.; Gao, Y.; Shi, L.; Liu, C.-X. Functional enzyme-based approach for linking microbial community functions with biogeochemical process kinetics. Environ. Sci. Technol. 2017, 51, 11848–11857. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Williams, A.J. Open science for identifying ‘known unknown’ chemicals. Environ. Sci. Technol. 2017, 51, 5357–5359. [Google Scholar] [CrossRef]
- Seviour, R.J.; Nielsen, P.H. Microbial Ecology of Activated Sludge; IWA Publishing: London, UK, 2010. [Google Scholar]
- Duportet, X.; Aggio, R.B.M.; Carneiro, S.; Villas-Bôas, S.G. The biological interpretation of metabolomic data can be misled by the extraction method used. Metabolomics 2012, 8, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Pinu, F.R.; Villas-Boas, S.G.; Aggio, R. Analysis of intracellular metabolites from microorganisms: Quenching and extraction protocols. Metabolites 2017, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Villas-Bôas, S.G.; Højer-Pedersen, J.; Akesson, M.; Smedsgaard, J.; Nielsen, J. Global metabolite analysis of yeast: Evaluation of sample preparation methods. Yeast 2005, 22, 1155–1169. [Google Scholar] [CrossRef] [Green Version]
- Herold, M.; Arbas, S.M.; Narayanasamy, S.; Muller, E.E.L.; Kleine-Borgmann, L.; Lebrun, L.A.; Kunath, B.; Roume, H.; Thieme, A.; Bessarab, I.; et al. Integration of time-series meta-omics data reveals how microbial ecosystems respond to disturbance. Nat. Commun. 2020, 11, 5281. [Google Scholar] [CrossRef]
- Cydzik-Kwiatkowska, A.; Zielińska, M. Bacterial communities in full-scale wastewater treatment systems. World J Microbiol. Biotechnol. 2016, 32, 66. [Google Scholar] [CrossRef] [Green Version]
- Narayanasamy, S.; Muller, E.E.L.; Sheik, A.R.; Wilmes, P. Integrated omics for the identification of key functionalities in biological wastewater treatment microbial Communities. Microb. Biotechnol. 2015, 8, 363–368. [Google Scholar] [CrossRef]
- Oehmen, A.; Lemos, P.C.; Carvalho, G.; Yuan, Z.; Keller, J.; Blackall, L.L.; Reis, M.A.M. Advances in enhanced biological phosphorus removal: From micro to macro scale. Water Res. 2007, 41, 2271–2300. [Google Scholar] [CrossRef]
- Parsons, S.A.; Smith, J.A. Phosphorus removal and recovery from municipal wastewaters. Elements 2008, 4, 109–112. [Google Scholar] [CrossRef]
- Zou, H.; Wang, Y. Phosphorus removal and recovery from domestic wastewater in a novel process of enhanced biological phosphorus removal coupled with crystallization. Bioresour. Technol. 2016, 211, 87–92. [Google Scholar] [CrossRef]
- Tomei, M.C.; Stazi, V.; Daneshgar, S.; Capodaglio, A.G. Holistic approach to phosphorus recovery from urban wastewater: Enhanced biological removal combined with precipitation. Sustain. Sci. Pract. Policy 2020, 12, 575. [Google Scholar] [CrossRef] [Green Version]
- Law, Y.Y.; Kirkegaard, R.H.; Cokro, A.A.; Liu, X.; Arumugam, K.; Xie, C.; Stokholm-Bjerregaard, M.; Drautz-Moses, D.I.; Nielsen, P.H.; Wuertz, S.; et al. Integrative microbial community analysis reveals full-scale enhanced biological phosphorus removal under tropical conditions. Sci. Rep. 2016, 6, 25719. [Google Scholar] [CrossRef]
- Nielsen, P.H.; McIlroy, S.J.; Albertsen, M.; Nierychlo, M. Re-evaluating the microbiology of the enhanced biological phosphorus removal process. Curr. Opin. Biotechnol. 2019, 57, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Stokholm-Bjerregaard, M.; McIlroy, S.J.; Nierychlo, M.; Søren, M.; Albertsen, M.; Nielsen, P.H. A critical assessment of the microorganisms proposed to be important to enhanced biological phosphorus removal in full-scale wastewater treatment systems. Front. Microbiol. 2017, 8, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skennerton, C.T.; Barr, J.J.; Slater, F.R.; Bond, P.L.; Tyson, G.W. Expanding our view of genomic diversity in Candidatus Accumulibacter clades. Environ. Microbiol. 2015, 17, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, K.; Bağcı, C.; Bessarab, I.; Beier, S.; Buchfink, B.; Górska, A.; Qiu, G.; Huson, D.H.; Williams, R.B.H. Annotated bacterial chromosomes from frame-shift-corrected long-read metagenomic data. Microbiome 2019, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Qiu, G.-L.; Liu, X.-H.; Saw, N.M.M.T.; Law, Y.Y.; Zuniga-Montanez, R.E.; Thi, S.S.; Nguyen, T.Q.N.; Nielsen, P.N.; Williams, R.B.H.; Wuertz, S. Metabolic traits of Accumulibacter Clade IIF Strain SCELSE-1 using amino acids as carbon sources for enhanced biological phosphorus removal. Environ. Sci. Technol. 2020, 54, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Nielsen, J.L.; Nielsen, P.H. Identity and ecophysiology of uncultured actinobacterial polyphosphate-accumulating organisms in full-scale enhanced biological phosphorus removal plants. Appl Environ Microbiol. 2005, 71, 4076–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Qiu, G.L.; Zuniga-Montanez, R.E.; Williams, R.B.H.; Wuertz, S. Recent advances in understanding the ecophysiology of enhanced biological phosphorus removal. Curr. Opin. Biotechnol. 2021, 67, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Daims, H.; Taylor, M.W.; Wagner, M. Wastewater treatment: A model system for microbial ecology. Trends Biotechnol. 2006, 24, 483–489. [Google Scholar] [CrossRef]
- Anderson, M.; Ter Braak, C.J.F. Permutation tests for multi-factorial analysis of variance. J. Stat. Comput. Simul. 2003, 73, 85–113. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA); Balakrishnan, N., Colton, T., Everitt, B., Piegorsch, W., Ruggeri, F., Teugels, J.L., Eds.; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Kim, H.K.; Verpoorte, R. Sample preparation for plant metabolomics. Phytochem. Anal. 2010, 21, 4–13. [Google Scholar] [CrossRef]
- Maharjan, R.P.; Ferenci, T. Global metabolite analysis: The influence of extraction methodology on metabolome profiles of Escherichia coli. Anal. Biochem. 2003, 313, 145–154. [Google Scholar] [CrossRef]
- Dettmer, K.; Nürnberger, N.; Kaspar, H.; Gruber, M.A.; Almstetter, M.F.; Oefner, P.J. Metabolite extraction from adherently growing mammalian cells for metabolomics studies: Optimization of harvesting and extraction protocols. Anal. Bioanal. Chem. 2011, 399, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Kimball, E.; Rabinowitz, J.D. Identifying decomposition products in extracts of cellular metabolites. Anal. Biochem. 2006, 358, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamerly, T.; Tripet, B.P.; Tigges, M.; Giannone, R.J.; Wurch, L.; Hettich, R.L.; Podar, M.; Copié, V.; Bothner, B. Untargeted metabolomics studies employing NMR and LC-MS reveal metabolic coupling between Nanoarcheum equitans and its archaeal host Ignicoccus hospitalis. Metabolomics 2015, 11, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Villas-Bôas, S.G.; Bruheim, P. Cold glycerol-saline: The promising quenching solution for accurate intracellular metabolite analysis of microbial cells. Anal. Biochem. 2007, 370, 87–97. [Google Scholar] [CrossRef]
- Seviour, T.; Derlon, N.; Dueholm, M.S.; Flemming, H.C.; Girbal-Neuhauser, E.; Horn, H.; Kjelleberg, S.; van Loosdrecht, M.C.M.; Lotti, T.; Malpei, M.F.; et al. Extracellular polymeric substances of biofilms: Suffering from an identity crisis. Water Res. 2019, 151, 1–7. [Google Scholar] [CrossRef]
- Tipthara, P.; Kunacheva, C.; Soh, Y.N.A.; Wong, S.C.C.; Ng, S.P.; Stuckey, D.C.; Boehm, B.O. Global profiling of metabolite and lipid soluble microbial products in anaerobic wastewater reactor supernatant using UPLC-MSE. J. Proteome Res. 2017, 16, 559–570. [Google Scholar] [CrossRef]
- Roume, H.; Heintz-Buschart, A.; Muller, E.E.L.; Wilmes, P. Sequential isolation of metabolites, RNA, DNA and proteins from the same unique sample. Methods Enzymol. 2013, 531, 219–236. [Google Scholar]
- Roy Chowdhury, T.; Lee, J.Y.; Bottos, E.M.; Brislawn, C.J.; White, R.A., 3rd; Bramer, L.M.; Brown, J.; Zucker, J.D.; Kim, Y.M.; Jumpponen, A.; et al. Metaphenomic responses of a native prairie soil microbiome to moisture perturbations. mSystems 2019, 4, e00061-19. [Google Scholar] [CrossRef] [Green Version]
- Swenson, T.L.; Northen, T.R. Untargeted soil metabolomics using liquid chromatography-mass spectrometry and gas chromatography-mass spectrometry. Methods Mol. Biol. 2019, 1859, 97–109. [Google Scholar]
- Stewart, E.J. Growing Unculturable bacteria. J. Bacteriol. 2012, 194, 4151–4160. [Google Scholar] [CrossRef] [Green Version]
- Barker, D.J.; Stuckey, D.C. A review of soluble microbial products (SMP) in wastewater treatment systems. Water Res. 1999, 33, 3063–3082. [Google Scholar] [CrossRef]
- Helmus, R.; Ter Laak, T.L.; van Wezel, A.P.; de Voogt, P.; Schymanski, E. patRoon: Open source software platform for environmental mass spectrometry based non-target screening. J. Chemoinformatics 2021, 13, 1. [Google Scholar]
- Schlegel, H.G.; Jannasch, H.W. Enrichment cultures. Annu. Rev. Microbiol. 1967, 21, 49–70. [Google Scholar] [CrossRef]
- Lu, H.; Oehmen, A.; Virdis, B.; Keller, J.; Yuan, Z. Obtaining highly enriched cultures of Candidatus Accumulibacter phosphates through alternating carbon sources. Water Res. 2006, 40, 3838–3848. [Google Scholar] [CrossRef]
- Smolders, G.J.; van der Meij, J.; van Loosdrecht, M.C.; Heijnen, J.J. Model of the anaerobic metabolism of the biological phosphorus removal process: Stoichiometry and pH influence. Biotechnol. Bioeng. 1994, 43, 461–470. [Google Scholar] [CrossRef]
- Vrhovsek, U.; Masuero, D.; Gasperotti, M.; Franceschi, P.; Caputi, L.; Viola, R.; Mattivi, F. A versatile targeted metabolomics method for the rapid quantification of multiple classes of phenolics in fruits and beverages. J. Agric. Food Chem. 2012, 60, 8831–8840. [Google Scholar] [CrossRef]
- Lewis, M.R.; Pearce, J.T.M.; Spagou, K.; Green, M.; Dona, A.C.; Yuen, A.H.Y.; David, M.; Berry, D.J.; Chappell, K.; Horneffer-van der Sluis, V.; et al. Development and application of ultra-performance liquid chromatography-TOF MS for precision large scale urinary metabolic phenotyping. Anal. Chem. 2016, 88, 9004–9013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorkas, P.A.; Isaac, G.; Anwar, M.A.; Davies, A.H.; Want, E.J.; Nicholson, J.K.; Holmes, E. Untargeted UPLC-MS profiling pipeline to expand tissue metabolome coverage: Application to cardiovascular disease. Anal. Chem. 2015, 87, 4184–4193. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.r-project.org/ (accessed on 15 March 2021).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 15 March 2021)ISBN 978-3-319-24277-4.
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 2019 35, 526–528. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.D.; Wiemann, S. KEGGgraph: A graph approach to KEGG PATHWAY in R and Bioconductor. Bioinformatics 2009, 25, 1470–1471. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, G.; Zuniga-Montanez, R.; Law, Y.; Thi, S.S.; Nguyen, T.Q.N.; Eganathan, K.; Liu, X.; Nielsen, P.H.; Williams, R.B.H.; Wuertz, S. Polyphosphate-accumulating organisms in full-scale tropical wastewater treatment plants use diverse carbon sources. Water Res. 2019, 149, 496–510. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode | Reactor | PERMANOVA Term | DF | SS | MS | Pseudo_F | p-Value |
|---|---|---|---|---|---|---|---|
| Positive | A | Solvent | 4 | 28,744 | 7186 | 45 | 0.0001 |
| Time | 3 | 2513 | 838 | 3.8 | 0.0034 | ||
| Solvent × Time | 12 | 6085 | 507 | 3.2 | 0.0003 | ||
| Residuals | 32 | 5084 | 159 | --- | --- | ||
| Positive | B | Solvent | 2 | 5399 | 2699 | 140 | 0.0001 |
| Type | 1 | 36,844 | 36,844 | 1753 | 0.0002 | ||
| Time | 1 | 85 | 85 | 4.5 | 0.0334 | ||
| Solvent × Type | 2 | 3965 | 1983 | 121 | 0.0001 | ||
| Solvent × Time | 2 | 42 | 21 | 1.1 | 0.3918 | ||
| Type × Time | 1 | 88 | 88 | 4.2 | 0.0251 | ||
| Solvent × Type × Time | 2 | 42 | 31 | 1.9 | 0.0948 | ||
| Residuals | 7 | 115 | 16 | --- | --- | ||
| Negative | A | Solvent | 4 | 28,884 | 7221 | 61 | 0.0001 |
| Time | 3 | 2180 | 727 | 7.9 | 0.0009 | ||
| Solvent × Time | 12 | 5894 | 491 | 4.2 | 0.0001 | ||
| Residuals | 32 | 3777 | 118 | --- | --- | ||
| Negative | B | Solvent | 2 | 6181 | 3091 | 184 | 0.0001 |
| Type | 1 | 47,733 | 47,733 | 2325 | 0.0001 | ||
| Time | 1 | 80 | 80 | 4.1 | 0.0333 | ||
| Solvent × Type | 2 | 6906 | 3453 | 198 | 0.0001 | ||
| Sovent × Time | 2 | 44 | 22 | 1.3 | 0.1800 | ||
| Type × Time | 1 | 91 | 91 | 4.4 | 0.0196 | ||
| Solvent × Type × Time | 2 | 36 | 18 | 1.0 | 0.4429 | ||
| Residuals | 7 | 112 | 17 | --- | --- |
| Solvent | Total Number of Detected Peaks | Total Number of Detected Compounds | ||||||
|---|---|---|---|---|---|---|---|---|
| Reactor A | Reactor B a | Reactor A | Reactor B a | |||||
| Positive Mode | Negative Mode | Positive Mode | Negative Mode | Positive Mode | Negative Mode | Positive Mode | Negative Mode | |
| M b | 6934 | 2804 | 1262 | 1428 | 3025 | 1429 | 957 | 928 |
| MW1 c | 8183 | 4045 | 1334 | 1816 | 3188 | 1770 | 1058 | 1104 |
| MW2 d | 7699 | 3852 | --- | --- | 3142 | 1731 | --- | --- |
| MW3 e | 7941 | 3604 | --- | --- | 3166 | 1697 | --- | --- |
| MCW_M f | 7825 | 3702 | 1364 | 1735 | 3143 | 1729 | 1024 | 1067 |
| MCW_C g | 2595 | 529 | 3507 | 514 | 641 | 68 | 1013 | 117 |
| Solvent | Total Number of Significant Mass Features (m/z) | Total Number of Detected Compounds | ||||||
|---|---|---|---|---|---|---|---|---|
| Reactor A | Reactor B | Reactor A | Reactor B | |||||
| Positive Mode | Negative Mode | Positive Mode | Negative Mode | Positive Mode | Negative Mode | Positive Mode | Negative Mode | |
| M a | 126 | 319 | 740 | 1086 | 112 | 275 | 483 | 782 |
| MW1 b | 1334 | 719 | 1003 | 1643 | 460 | 259 | 761 | 1042 |
| MW2 c | 6124 | 2325 | --- | --- | 2763 | 1246 | --- | --- |
| MW3 d | 35 | 431 | --- | --- | 2538 | 398 | --- | --- |
| MCW_M e | 10 | 0 | 857 | 1488 | 2 | 0 | 625 | 946 |
| MCW_C f | 0 | 0 | 3335 | 501 | 0 | 0 | 878 | 93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saw, N.M.M.T.; Suwanchaikasem, P.; Zuniga-Montanez, R.; Qiu, G.; Marzinelli, E.M.; Wuertz, S.; Williams, R.B.H. Influence of Extraction Solvent on Nontargeted Metabolomics Analysis of Enrichment Reactor Cultures Performing Enhanced Biological Phosphorus Removal (EBPR). Metabolites 2021, 11, 269. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11050269
Saw NMMT, Suwanchaikasem P, Zuniga-Montanez R, Qiu G, Marzinelli EM, Wuertz S, Williams RBH. Influence of Extraction Solvent on Nontargeted Metabolomics Analysis of Enrichment Reactor Cultures Performing Enhanced Biological Phosphorus Removal (EBPR). Metabolites. 2021; 11(5):269. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11050269
Chicago/Turabian StyleSaw, Nay Min Min Thaw, Pipob Suwanchaikasem, Rogelio Zuniga-Montanez, Guanglei Qiu, Ezequiel M. Marzinelli, Stefan Wuertz, and Rohan B. H. Williams. 2021. "Influence of Extraction Solvent on Nontargeted Metabolomics Analysis of Enrichment Reactor Cultures Performing Enhanced Biological Phosphorus Removal (EBPR)" Metabolites 11, no. 5: 269. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11050269