
A Compositional Model to Predict the Aggregated Isotope Distribution for Average DNA and RNA Oligonucleotides

 , , ,
, , ,
Abstract
:
1. Introduction
2. Material and Methods
2.1. Experimental Procedures
2.2. Theoretical Data
2.3. Compositional Data Transformation
2.4. Modelling Approach
2.5. Prediction of the Isotopic Envelope
2.6. The Goodness-of-Fit Statistic
3. Results
3.1. Generation of the Data Sets
3.2. Model Selection and Training of the Model
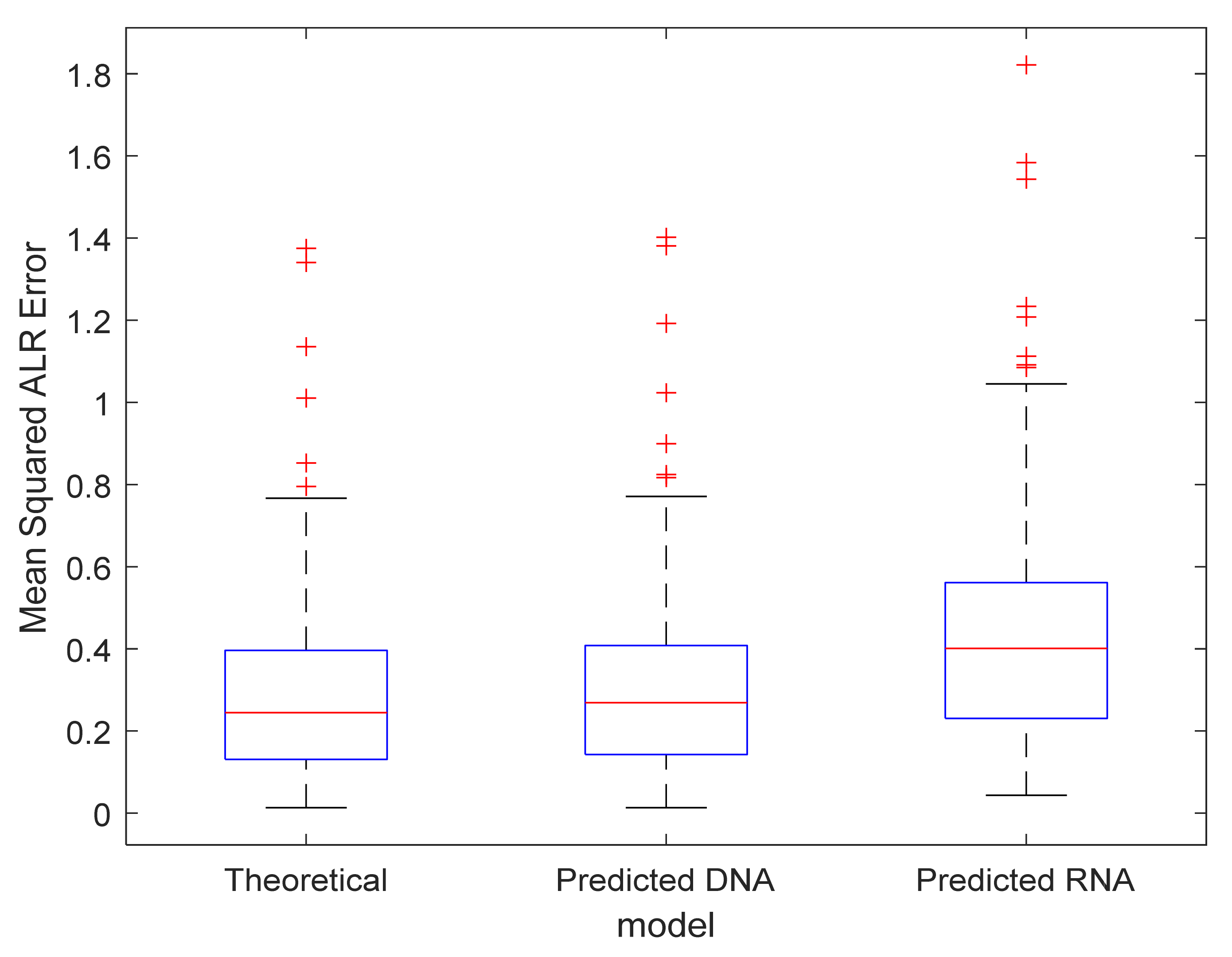
3.3. Exploration of the Proposed Error Metrics
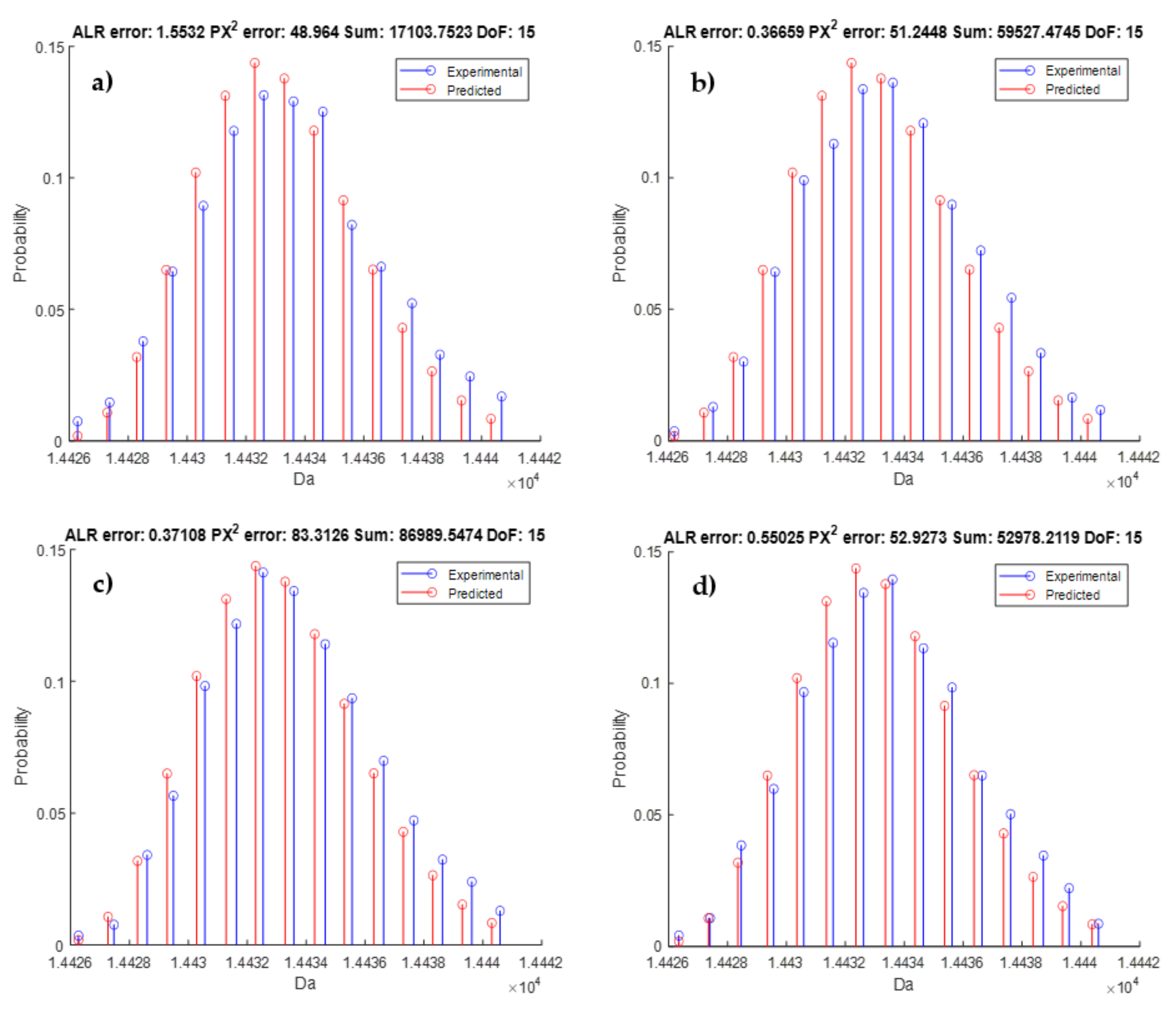
3.4. Model Validation
3.5. Software
4. Discussion
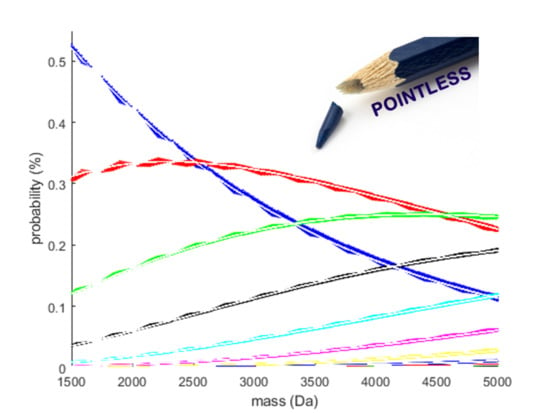
- Model fit: A polynomial model of order 10, which minimises the MSE on our test set, is acceptable given the vast amount of data points, making the risk of overfitting minimal. However, a few remarks are worth considering. Figure 2 illustrates that the density of data points increases with molecular weight. Since it is beneficial for a regression model to spend its flexibility in the dense data regions to minimise the MSE over the entire dataset, less flexibility remains for modelling the lower mass region. This effect is partly remediated by using a weighted least square regression approach that employs squared residuals as the weight. However, even these weighted errors do not contribute much to the overall MSE, leading to a biased model fit for the RNA model, as can be seen in panel (b) of the Supplementary Figure S7. Furthermore, polynomial models with a high order are known to have irregular behaviour at the boundaries. In order to rectify this effect, we have used the full range dataset but restricted the prediction model to the range specified earlier. In future work, we will propose a model that is capable of handling these boundary effects and that can spread its flexibility more evenly over the data range even though the data is distributed in an unbalanced manner.
- Transformation of the simplex: The additive log-ratio transformation is the obvious choice for our modelling exercise since it can handle partially observed data. Here we opted for the monoisotopic variant as a reference. From Figure 2 it can be observed that the probability of this variant decreases rapidly with increasing molecular weight. For the current mass range, the division by small numbers remains within machine precision, but it can be argued that, for higher molecular weights above 30 kDa, a different reference has to be used to keep the transformation well-conditioned.
- Transformation of the observed isotope distribution: The benefit of the ALR transformation is that we can transform the partially observed data into ALR space and execute the goodness-of-fit comparison directly with the predicted ALR transformed isotopes. This strategy is only possible if the monoisotopic variant has a quantifiable intensity in an observed spectrum. Two remarks are worth considering. Firstly, as mentioned in the previous bullet point, the monoisotopic variant falls below the limit of detection for large molecules, obstructing the transformation of the observed isotope distribution in the ALR space. Secondly, a large error on the monoisotopic intensity will propagate severely in the ALR error as this reference is used to transform all the isotopes. To remediate this effect, we could argue to divide the mass range into distinct bins for which an optimal reference is chosen.
- Multinomial test: In order to relax the dependency of the goodness-of-fit statistic on the monoisotopic variant, we proposed a score that uses the back-transformed probabilities. This score can be computed on a partially observed isotope distribution, provided that the monoisotopic mass is known and the user has knowledge of which isotopes are observed. Another remark is that the multinomial test is approximated by the Pearson’s chi-squared test. However, our implementation does not rely on formal statistics. Furthermore, instead of the actual number of ions, the intensity values are used as a proxy. Therefore, the proposed mean Pearson’s chi-squared error just tries to quantify the goodness-of-fit. As discussed earlier, this metric is able to tolerate larger error deviations when intensities are low (i.e., low ion statistics) or can be more stringent for high intensities (i.e., high ion statistics). The latter states that a one-to-one relation between intensity and ion statistics exists, but the kind of relation depends on the instrument type. Hence, without this formal testing framework, the MPCSE is not interoperable between platforms. Therefore, we suggest calibrating this metric on every instrument using a benchmark dataset for optimal thresh old selection.
- Choice of the model covariate: The monoisotopic mass is used as the predictor variable in our model (x-axis in Figure 2 and Figure 3). Hence, to forecast the isotope distribution we need to have the monoisotopic mass as an input. From earlier discussion, we know that the monoisotopic variant is not observed for high molecular weight molecules. Three procedures can be proposed to solve this conundrum. Firstly, the same model can be devised but with a different covariate, for example, the average or most abundant mass of the molecules. Secondly, a model similar in spirit as MIND by Lermyte et al. [35] can be proposed for DNA that predicts the monoisotopic mass based on the partially observed isotope distribution. Thirdly, we can use the model in combination with the fitting procedure of Senko et al. [36] that scales the elemental composition of an average amino acid, denoted as averagine. However, for the use case in nucleic acids, this would require the counterpart of the averagine model for DNA and RNA.
- Univariate approach: From the residual plot on the predicted probabilities and our ad hoc method to compute the centroid masses, we can observe different types of correlation in the data. It is worthwhile to investigate whether this correlation can be exploited in a multivariate analysis in order to obtain a better prediction model. On the other hand, we have demonstrated that current predictions of probability and mass are very close to the actual values, and that the errors are ignorable given current instrument precision and mass accuracy.
5. Concluding Remark
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banoub, J.H.; Newton, R.P.; Esmans, E.; Ewing, D.F.; Mackenzie, G. Recent Developments in Mass Spectrometry for the Characterization of Nucleosides, Nucleotides, Oligonucleotides, and Nucleic Acids. Chem. Rev. 2005, 105, 1869–1916. [Google Scholar] [CrossRef]
- Limbach, P.A. Indirect mass spectrometric methods for characterizing and sequencing oligonucleotides. Mass Spectrom. Rev. 1996, 15, 297–336. [Google Scholar] [CrossRef]
- Schürch, S. Characterization of nucleic acids by tandem mass spectrometry—The second decade (2004–2013): From DNA to RNA and modified sequences. Mass Spectrom. Rev. 2014, 35, 483–523. [Google Scholar] [CrossRef]
- Wein, S.; Andrews, B.; Sachsenberg, T.; Santos-Rosa, H.; Kohlbacher, O.; Kouzarides, T.; Garcia, B.A.; Weisser, H. A computational platform for high-throughput analysis of RNA sequences and modifications by mass spectrometry. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.K.; Glick, J.; Liao, Q.; Shen, C.; Vouros, P. GenoMass software: A tool based on electrospray ionization tandem mass spectrometry for characterization and sequencing of oligonucleotide adducts. J. Mass Spectrom. 2012, 47, 490–501. [Google Scholar] [CrossRef] [Green Version]
- Sample, P.J.; Gaston, K.W.; Alfonzo, J.D.; Limbach, P.A. RoboOligo: Software for mass spectrometry data to support manual and de novo sequencing of post-transcriptionally modified ribonucleic acids. Nucleic Acids Res. 2015, 43, e64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tretyakova, N.; Villalta, P.; Kotapati, S. Mass Spectrometry of Structurally Modified DNA. Chem. Rev. 2013, 113, 2395–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giessing, A.M.; Kirpekar, F. Mass spectrometry in the biology of RNA and its modifications. J. Proteom. 2012, 75, 3434–3449. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, C.; Limbach, P.A. Mass spectrometry of modified RNAs: Recent developments. Analyst 2016, 141, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Hagelskamp, F.; Borland, K.; Ramos, J.; Hendrick, A.G.; Fu, D.; Kellner, S. Broadly applicable oligonucleotide mass spectrometry for the analysis of RNA writers and erasers in vitro. Nucleic Acids Res. 2020, 48, e41. [Google Scholar] [CrossRef] [PubMed]
- Thüring, K.; Schmid, K.; Keller, P.; Helm, M. Analysis of RNA modifications by liquid chromatography–tandem mass spectrometry. Methods 2016, 107, 48–56. [Google Scholar] [CrossRef]
- Zhang, N.; Shi, S.; Jia, T.Z.; Ziegler, A.; Yoo, B.; Yuan, X.; Li, W.; Zhang, S. A general LC-MS-based RNA sequencing method for direct analysis of multiple-base modifications in RNA mixtures. Nucleic Acids Res. 2019, 47, e125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourshahian, S. Therapeutic Oligonucleotides, Impurities, Degradants, and Their Characterization by Mass Spectrometry. Mass Spectrom. Rev. 2021, 40, 75–109. [Google Scholar] [CrossRef]
- Capaldi, D.; Akhtar, N.; Atherton, T.; Benstead, D.; Charaf, A.; De Vijlder, T.; Heatherington, C.; Hoernschemeyer, J.; Jiang, H.; Rieder, U.; et al. Strategies for Identity Testing of Therapeutic Oligonucleotide Drug Substances and Drug Products. Nucleic Acid Ther. 2020, 30, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Sharma, I.; Glick, J. The expanding role of mass spectrometry in the field of vaccine development. Mass Spectrom. Rev. 2018, 39, 83–104. [Google Scholar] [CrossRef]
- Poveda, C.; Biter, A.B.; Bottazzi, M.E.; Strych, U. Establishing Preferred Product Characterization for the Evaluation of RNA Vaccine Antigens. Vaccines 2019, 7, 131. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Yu, N.; Kim, J.; Murgo, J.-R.; Kissai, M.; Ravichandran, K.R.; Miracco, E.J.; Presnyak, V.; Hua, S. Oligonucleotide Sequence Mapping of Large Therapeutic mRNAs via Parallel Ribonuclease Digestions and LC-MS/MS. Anal. Chem. 2019, 91, 8500–8506. [Google Scholar] [CrossRef] [PubMed]
- Valkenborg, D.; Jansen, I.; Burzykowski, T. A model-based method for the prediction of the isotopic distribution of peptides. J. Am. Soc. Mass Spectrom. 2008, 19, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Valkenborg, D.; Mertens, I.; Lemière, F.; Witters, E.; Burzykowski, T. The isotopic distribution conundrum. Mass Spectrom. Rev. 2011, 31, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Böcker, S.; Letzel, M.C.; Lipták, Z.; Pervukhin, A. SIRIUS: Decomposing isotope patterns for metabolite identification†. Bioinformatics 2008, 25, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Dittwald, P.; Claesen, J.; Burzykowski, T.; Valkenborg, D.; Gambin, A. BRAIN: A Universal Tool for High-Throughput Calculations of the Isotopic Distribution for Mass Spectrometry. Anal. Chem. 2013, 85, 1991–1994. [Google Scholar] [CrossRef]
- Dittwald, P.; Valkenborg, D. BRAIN 2.0: Time and Memory Complexity Improvements in the Algorithm for Calculating the Isotope Distribution. J. Am. Soc. Mass Spectrom. 2014, 25, 588–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łącki, M.K.; Startek, M.; Valkenborg, D.; Gambin, A. IsoSpec: Hyperfast Fine Structure Calculator. Anal. Chem. 2017, 89, 3272–3277. [Google Scholar] [CrossRef]
- Coursey, J.S.; Schwab, D.J.; Tsai, J.J.; Dragoset, R.A. Atomic Weights and Isotopic Compositions (Version 4.1); National Institute of Standards and Technology: Gaithersburg, MD, USA, 2015.
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Gatto, L.; Gibb, S.; Rainer, J. MSnbase, Efficient and Elegant R-Based Processing and Visualization of Raw Mass Spectrometry Data. J. Proteome Res. 2021, 20, 1063–1069. [Google Scholar] [CrossRef]
- Gatto, L.; Lilley, K.S. MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 2011, 28, 288–289. [Google Scholar] [CrossRef] [Green Version]
- Yergey, J.A. A general approach to calculating isotopic distributions for mass spectrometry. Int. J. Mass Spectrom. Ion Phys. 1983, 52, 337–349. [Google Scholar] [CrossRef]
- Aitchison, J. The Statistical Analysis of Compositional Data. J. R. Stat. Soc. Ser. B 1982, 44, 139–177. [Google Scholar] [CrossRef]
- Aitchison, J. The Statistical Analysis of Compositional Data; Chapman and Hall: London, UK, 1986. [Google Scholar]
- Aitchison, J. Principles of compositional data analysis. Inst. Math. Stat. Collect. 1994, 24, 73–81. [Google Scholar]
- Aitchison, J.; Shen, S.M. Logistic-Normal Distributions: Some Properties and Uses. Biometrika 1980, 67, 261–272. [Google Scholar] [CrossRef]
- Aitchison, J.; Barceló-Vidal, C.; Martín-Fernández, J.A.; Pawlowsky-Glahn, V. Logratio Analysis and Compositional Distance. Math. Geol. 2000, 32, 271–275. [Google Scholar] [CrossRef]
- Pearson, K.X. On the criterion that a given system of deviations from the probable in the case of a correlated system of variables is such that it can be reasonably supposed to have arisen from random sampling. London Edinb. Dublin Philos. Mag. J. Sci. 1900, 50, 157–175. [Google Scholar] [CrossRef] [Green Version]
- Lermyte, F.; Dittwald, P.; Claesen, J.; Baggerman, G.; Sobott, F.; O’Connor, P.B.; Laukens, K.; Hooyberghs, J.; Gambin, A.; Valkenborg, D. MIND: A Double-Linear Model to Accurately Determine Monoisotopic Precursor Mass in High-Resolution Top-Down Proteomics. Anal. Chem. 2019, 91, 10310–10319. [Google Scholar] [CrossRef] [PubMed]
- Senko, M.W.; Beu, S.C.; McLaffertycor, F.W. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass Spectrom. 1995, 6, 229–233. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | DNA_SHORT1 | DNA_SHORT2 |
| Type | DNA | DNA |
| Sequence | GCC ACA TAT GAG AGT GGA TTT GTC ATT | GGT GCC CCA GAA TCT CTC AGC CT |
| Elemental formula | C266H334N100O162P26 | C221H282N82O137P22 |
| Monoisotopic mass | 8325.41493 | 6957.184784 |
| Charge states | 6 to 12 | 5 to 9 |
| Elution ranges | 10.95 min–11.05 min (10 scans) | 10.32 min–10.45 min (14 scans) |
| Replicates | 7 × 10 = 70 | 5 × 14 = 70 |
| Name | RNA-like | DNA_long |
| Type | RNA | DNA |
| Sequence | As-Afs-Cs-Af-U-Uf-G-A-G-Cf-G-Af-U-Af-U-Cf-C-As-C [N = 2′OMe, Nf = 2′F, s or PS, phosphorothioate. PO, phosphodiester] | GAG ATC TCT GCT TCT GAT GGC TCT CTG GTT ACT GCC AGT TGA ATC TG |
| Elemental formula | C192H239O117N73P18S4F8 | C459H582N162O290P46 |
| Monoisotopic mass | 6275.90281 | 14,426.37043 |
| Charge states | 5 to 8 | 15 to 18 |
| Elution ranges | 13.75 min–13.85 min (12 scans) | 12.1 min–12.3 min (23 scans) |
| Replicates | 4 × 12 = 48 | 4 × 23 = 92 |
| DNA | RNA | ||||||
|---|---|---|---|---|---|---|---|
| Isotope | Mass Difference (Da) | Isotope | Mass Difference (Da) | Isotope | Mass Difference (Da) | Isotope | Mass Difference (Da) |
| 1 | 0 | 11 | 10.02608399 | 1 | 0 | 11 | 10.02587038 |
| 2 | 1.002707 | 12 | 11.02860871 | 2 | 1.002698922 | 12 | 11.02836784 |
| 3 | 2.005384 | 13 | 12.03112309 | 3 | 2.005361437 | 13 | 12.03085481 |
| 4 | 3.008035 | 14 | 13.03362787 | 4 | 3.007994300 | 14 | 13.03333209 |
| 5 | 4.010663 | 15 | 14.03612367 | 5 | 4.010602048 | 15 | 14.03580039 |
| 6 | 5.013272 | 16 | 15.03861108 | 6 | 5.013188104 | 16 | 15.03826032 |
| 7 | 6.015864 | 17 | 16.0410906 | 7 | 6.015755128 | 17 | 16.04071244 |
| 8 | 7.018439 | 18 | 17.0435627 | 8 | 7.018305257 | 18 | 17.04315722 |
| 9 | 8.021000 | 19 | 18.0460278 | 9 | 8.020840240 | 19 | 18.04559513 |
| 10 | 9.023548 | 20 | 19.04848627 | 10 | 9.023361538 | 20 | 19.04802655 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agten, A.; Prostko, P.; Geubbelmans, M.; Liu, Y.; De Vijlder, T.; Valkenborg, D. A Compositional Model to Predict the Aggregated Isotope Distribution for Average DNA and RNA Oligonucleotides. Metabolites 2021, 11, 400. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11060400
Agten A, Prostko P, Geubbelmans M, Liu Y, De Vijlder T, Valkenborg D. A Compositional Model to Predict the Aggregated Isotope Distribution for Average DNA and RNA Oligonucleotides. Metabolites. 2021; 11(6):400. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11060400
Chicago/Turabian StyleAgten, Annelies, Piotr Prostko, Melvin Geubbelmans, Youzhong Liu, Thomas De Vijlder, and Dirk Valkenborg. 2021. "A Compositional Model to Predict the Aggregated Isotope Distribution for Average DNA and RNA Oligonucleotides" Metabolites 11, no. 6: 400. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11060400